
Biochem.
J.
(1994)
298,
9-16
(Printed
in
Great
Britain)
Statistical
prediction
of
the
locus
of
endoproteolytic
cleavage
of
the
nascent
polypeptide
in
glycosylphosphatidylinositol-anchored
proteins
A§ok
C.
ANTONY*t
and
Michael
E.
MILLERt
TDivision
of
Hematology-Oncology,
Department
of
Medicine,
Indiana
University
School
of
Medicine,
Indianapolis,
IN
46202-5121,
U.S.A.,
and
tSection
on
Biostatistics,
Department
of
Public
Health
Sciences,
Bowman
Gray
School
of
Medicine,
Winston-Salem,
NC
27157-1063,
U.S.A.
Existing
methods
of
identifying
the
cleavage
site
of
the
nascent
polypeptide
and
the
C-terminal
residue
to
which
the
glycosyl-
phosphatidylinositol
(GPI)
anchor
is
attached
in
mature
GPI-
anchored
proteins
are
technically
difficult
and
labour-intensive.
We
tested
the
hypothesis
that
it
was
possible
to
predict
this
locus
using
data
from
the
cDNA-deduced
amino
acid
sequence
and
amino
acid
composition
of
GPI-anchored
proteins.
We
employed
a
statistical
approach
which
allowed
repeated
x2
comparisons
between
the
proportions
of
residual
amino
acids
in
the
major
body
of
the
cDNA-deduced
polypeptide
(minus
the
N-terminal
signal
peptide)
after
repeated
computer-generated
progressive
exoproteolysis
from
its
C-terminus
one
amino
acid
at
a
time
and
the
fixed
proportion
of
amino
acids
obtained
from
amino
acid
analysis
of
the
mature
GPI-anchored
protein.
Initial
comparison
INTRODUCTION
Among
several
C-terminal
modifications
involving
proteins
des-
tined
for
membrane
insertion,
the
signal
for
farnesylation
forms
one
category
involving
deletion
of
the
penultimate
three
amino
acids
from
the
CAAX
box
sequence
preceded
by
addition
of
the
isoprenylated
fatty
acid
tail
[1].
In
contrast,
for
proteins
that
are
destined
for
insertion
into
the
membrane
via
glycosylphospha-
tidylinositol
(GPI)
anchors
[2-6],
there
is
cleavage
of
a
larger
segment
of
the
C-terminal
nascent
polypeptide
followed
by
en
bloc
addition
of
a
preformed
GPI
anchor
which
is
mediated
by
a
putative
GPI
transamidase
enzyme.
Compared
with
other
clearly
defined
signals
for
post-translational
processing
of
pro-
teins,
there
is
(as
yet),
strictly
no
consensus
sequence
which
predicts
whether
a
given
cDNA-deduced
amino
acid
sequence
contains
information
for
GPI-anchor
addition
[2-6].
Recent
studies
have
identified
general
requirements
for
GPI-anchor
addition
which
include
a
minimal
length
and
a
large
proportion
of
hydrophobic
amino
acids
for
this
C-terminal
segment.
Fur-
thermore,
there
is
a
cleavage/attachment
site
located
10-12
residues
N-terminal
to
the
hydrophobic
domain
consisting
of
specific
small
acceptor
amino
acids
(for
the
GPI
anchor)
followed
by
at
least
one,
but
generally
two,
other
small
amino
acids
C-
terminal
to
this
site
[7-11].
However,
there
are
several
membrane-
associated
proteins
that
possess
similar
features
within
their
C-terminal
polypeptide
ends
which
allow
for
their
insertion
into
the
membrane
as
polypeptide
anchors,
and
in
some
GPI-
anchored
species
such
as
the
CD16
receptor,
the
distinction
between
ultimate
anchoring
via
a
polypeptide
or
GPI
anchor
has
been
localized
to
substitution
of
a
single
amino
acid
within
this
C-terminal
fragment
[12,13].
A
simple
screening
test
employed
to
identify
GPI-anchoring
is
to
determine
if
the
protein
is
released
from
membranes
as
a
of
the
two
parameters
invariably
revealed
a
relatively
high
x2
statistic
which
progressively
lowered
to
a
minimum
point
at
which
the
amino
acid
proportions
of
progressively
exoproteo-
lysed
polypeptide
and
fixed
endoproteolysed
polypeptides
of
the
mature
GPI-anchored
protein
were
in
closest
agreement.
This
objectively
defined
and
unique
minimum
point
of
closest
agree-
ment
accurately
identified
the
locus
of
post-translational
endo-
proteolytic
cleavage
of
the
nascent
polypeptide
in
several
tissue-
specific
single-gene-encoded
GPI-anchored
proteins.
Thus
the
C-terminal
amino
acid
to
which
the
GPI
anchor
is
attached
can
be
rapidly
identified
using
data
from
the
cDNA
sequence
and
the
amino
acid
composition
of
proteins
suspected
to
be
GPI-anchored.
soluble
species
by
GPI-specific
phospholipase
C.
However,
as
there
are
some
GPI-linked
proteins
that
are
not
released
by
this
manoeuvre
[2-6,14],
this
test
is
not
infallible.
Differential
solubil-
ization
in
various
concentrations
of
Triton
X-1
14
and
phase
separation
at
the
cloud
point
of
Triton
X-1
14
has
also
been
recently
suggested
as
a
means
of
discriminating
GPI-anchored
from
transmembrane
polypeptide-anchored
proteins
[15],
but
cannot
provide
further
data
on
the
C-terminal
domains
that
are
determinants
of
membrane
association.
Another
general
method
of
identifying
whether
or
not
a
protein
is
GPI-anchored
involves
comparing
the
number
of
amino
acid
residues
of
the
protein
after
its
amino
acid
analysis
and
defining
whether
it
corresponds
in
size
to
the
cDNA-deduced
amino
acid
sequence
(minus
its
signal
sequence
which
would
not
be
represented
in
the
mature
protein).
However,
as
the
C-terminal
segment
corresponds
to
only
a
fraction
of
the
mature
polypeptide
[2-6],
this
method
of
subjective
evaluation
also
has
limitations.
In
addition,
deglycosylation
of
the
protein
and
SDS/PAGE
of
a
purified
GPI-anchored
protein
may
not
reveal
major
differences
in
Mr
compared
with
the
cDNA-deduced
polypeptide
(or
its
mRNA
translated
in
vitro
without
microsomes),
as
gel
electrophoresis
is
not
sensitive
enough
to
discriminate
loss
of
15-30
amino
acids
in
the
mature
GPI-anchored
polypeptide.
We
tested
the
hypothesis
that
the
locus
of
endoproteolytic
cleavage
of
the
nascent
polypeptide
to
which
the
GPI
anchor
is
attached
could
be
identified
on
the
basis
of
structural
charac-
teristics
of
the
cDNA
and
protein
(i.e.
information
on
the
cDNA
sequence
and
amino
acid
composition).
The
basis
for
this
hypothesis
exploited
the
fact
that
GPI-anchored
proteins
always
represent
truncated
forms
of
the
nascent
polypeptide.
We
have
validated
the
statistical
approach
and
provide
evidence
on
how
this
simple
objective
method
can
localize,
with
precision,
the
C-
terminal
amino
acid
to
which
the
GPI
anchor
is
attached;
this
Abbreviations
used:
GPI,
glycosylphosphatidylinositol;
PLAP,
placental
alkaline
phosphatase;
CEA,
carcinoembryonic
antigen;
DAF,
decay-
accelerating
factor;
VSG,
variant
surface
glycoprotein;
PARP,
procylic
acidic
repetitive
protein;
PFR,
placental
folate
receptor.
$
To
whom
correspondence
should
be
addressed.
Biochem.
J.
(1994)
298,
9-16
(Printed
in
Great
Britain)
9

10
A.
C.
Antony
and
M.
E.
Miller
method
can
also
identify
whether
a
protein
is,
in
fact,
likely
to
be
GPI-anchored
within
a
fraction
of
the
time
necessary
for
conventional
analytical
methods.
EXPERIMENTAL
Development
of
the
statistical
approach
We
hypothesized
that
there
should
be
a
mathematically
defined
and
consistent
relationship
between
the
proportions
of
the
various
amino
acids
in
the
nascent
polypeptide
of
a
GPI-anchored
protein
(taken
from
its
cDNA-deduced
amino
acid
sequence
minus
the
N-terminal
signal
peptide)
and
the
proportions
of
amino
acids
in
the
mature
protein
obtained
by
conventional
amino
acid
analysis.
Furthermore,
we
reasoned
that,
as
a
x2
goodness-of-fit
statistic
[16,17]
can
quantify
this
relationship,
we
might
be
able
to
define
the
precise
locus
of
post-translational
cleavage
of
the
C-terminus
of
the
nascent
polypeptide
(and
thereby
identify
the
precise
C-terminal
amino
acid
to
which
the
GPI
anchor
is
attached).
Some
discussion
of
the
use
of
the
x2
statistic
as
a
measure
of
distance
rather
than
mean
squared
difference
(another
popular
measure)
is
contained
in
Appendix
1.
A
simplified
diagram
illustrating
our
approach
is
shown
in
Figure
Al.
Initially,
we
calculated
a
x2
goodness-of-fit
statistic
to
de-
termine
how
closely
the
number
of
residues
from
the
amino
acid
analysis
agreed
with
those
observed
in
the
complete
cDNA-
deduced
amino
acid
sequence.
The
x2
goodness-of-fit
statistic
was
then
recalculated
N
times,
by
sequentially
removing
i
(i
=
1,...,N)
residues
from
the
C-terminus
of
the
cDNA-deduced
amino
acid
sequence
to
determine
if
this
could
provide
an
indication
of
how
much
of
the
sequenced
protein
had
been
left
out during
amino
acid
analysis
of
the
mature
protein
[17].
If
random
variability
does
not
exist
in
the
amino
acid
analysis
results,
then,
in
theory,
the
x2
statistic
will
equal
zero
immediately
after
removal
of
the
residue
at
which
the
nascent
polypeptide
is
truncated
before
addition
of
the
GPI
anchor.
However,
because
there
is
minor
random
variability
associated
with
production
of
amino
acid
data,
we
can
only
expect
the
goodness-of-fit
statistic
to
reach
a
local
minimum
at
the
point
at
which
the
proportions
must
closely
agree.
This
minimum
may
not
be
unique,
such
as
in
the
theoretical
situation
in
which
the
proportion
of
amino
acids
in
the
deleted
segment
comprising
the
C-terminal
sequence
exactly
matches
the
proportion
of
amino
acids
in
the
remainder
of
the
sequence.
This
is,
however,
unlikely
in
the
case
of
GPI-
anchored
proteins,
in
which
the
C-terminal
segment
destined
to
be
cleaved
from
the
mature
protein
comprises
(i)
only
a
small
fraction
of
the
complete
cDNA-predicted
sequence
and
(ii)
predominantly
hydrophobic
amino
acids,
which
contrasts
with
the
mixture
of
amino
acids
found
in
biologically
relevant
natural
proteins.
As
the
amino
acids
within
the
cDNA-deduced
N-
terminal
signal
sequence
would
not
be
found
in
the
mature
protein,
we
eliminated
the
amino
acids
of
the
signal
sequence
from
amino
acid
proportions
of
the
nascent
polypeptide.
GPI-anchored
proteins
subjected
to
analysis
We
studied
several
GPI-anchored
proteins
for
which
all
three
parameters
(cDNA
sequence,
amino
acid
analysis,
C-terminal
amino
acid
to
which
the
GPI
anchor
is
attached)
were
available.
These
included
Thy-I
[18,19],
variant
surface
glycoprotein
(VSG-
117)
from
Trypanosoma
brucei
[20,21],
placental
alkaline
phosphatase
(PLAP)
[22-24],
procyclic
acidic
repetitive
protein
(PARP)
from
T.
brucei
strain
427
and
TREU667
PARP
B-I
-a
well
as
carcinoembryonic
antigen
(CEA)
[28-30].
(For
CEA,
although
the
amino
acid
analysis
was
reported
in
mol
%,
the
total
number
of
amino
acid
residues
was
not
reported,
thus
compromising
our
ability
to
carry
out
direct
x2
comparisons.)
In
addition,
other
GPI-anchored
proteins
for
which
the
cDNA
sequence
and
the
precise
C-terminal
amino
acid
of
the
mature
protein
(GPI-anchor-attachment
site)
was
available,
but
for
which
the
amino
acid
composition
was
not
reported,
were
also
used
during
the
development
phases
of
this
study;
these
include
decay-accelerating
factor
(DAF)
[31,32]
and
5'-nucleotidase
[33,34].
An
example
of
the
statistical
program
for
analysis
of
VSG-1
17
is
provided
in
Appendix
2.
RESULTS
In
preliminary
studies,
we
calculated
the
value
of
the
x2
statistic
that
quantified
the
overall
'agreement'
between
the
proportions
of
amino
acids
in
the
nascent
polypeptide
(cDNA-deduced
amino
acid
sequence
minus
the
predicted
N-terminal
signal
peptide)
and
the
mature
523-amino
acid-long
protein
in
5'-nucleotidase
[33].
As
expected,
the
proportions
of
amino
acids
between
the
mature
and
nascent
5'-nucleotidase
polypeptide
initially
revealed
a
non-zero
x2
statistic
(equal
to
1.1270),
consistent
with
the
smaller
size
of
the
mature
protein
compared
with
the
nascent
polypeptide.
Next,
we
reasoned,
from
mathematical
principles,
that
the
x2
statistic
should
equal
zero
when
the
proportion
of
amino
acids
in
the
nascent
polypeptide
is
altered
such
that
there
is
no
difference
from
the
smaller-sized
(endoproteolytically
generated)
mature
protein;
at
issue
was
whether
the
x2
statistic
would
obtain
a
unique
zero
point.
This
was
verified
when
a
computer-generated
progressive
exoproteolysis
was
initiated
from
the
C-terminus
of
the
548-residue
nascent
polypeptide
one
amino
acid
at
a
time
with
repeated
monitoring
of
the
x2
statistic
[which
compared
the
exoproteolysed
nascent
polypeptide
(after
removal
of
each
amino
acid)
and
the
endoproteolysed
523-
residue
polypeptides
of
the
mature
protein].
We
observed
a
progressive
reduction
in
the
x2
statistic
value,
which
continued
with
progressive
exoproteolysis
of
the
nascent
polypeptide
until
it
reached
a
unique
trough
at
zero
when
the
proportions
of
amino
acids
in
the
exoproteolysed
polypeptide
were
the
same
as
those
in
the
endoproteolytically
cleaved
mature
protein.
Inter-
estingly,
this
zero
point
was
achieved
after
removal
of
only
25
amino
acid
residues
from
the
C-terminus
of
the
nascent
poly-
peptide.
This
locus
identified
Ser-523
as
the
C-terminal
amino
acid
in
the
exoproteolysed
polypeptide,
which
is
also
precisely
the
amino
acid
to
which
the
GPI
anchor
is
known
to
be
attached
in
mature
5'-nucleotidase
[34].
Further
computer-generated
exo-
proteolysis
beyond
this
zero
point
generated
a
smaller
poly-
peptide
than
the
endoproteolysed
polypeptide,
and
as
expected,
led
to
progressively
greater
x2
statistics.
It
should
be
pointed
out
that
the
lowest
x2
value
was
only
reached
once
during
this
exercise,
indicating
that
the
single
unique
lowest
point
was
unambiguous.
The
ability
to
obtain
a
unique
point
at
which
the
x2
statistic
equalled
zero
was
further
explored
after
additional
random
(theoretical)
endoproteolytic
cleavages
were
made
in
5'-
nucleotidase
to
remove
93,
158
and
223
amino
acid residues
from
the
C-terminus
of
the
nascent
polypeptide.
Again,
as
expected,
the
x2
statistic
reached
a
zero
point
only
when
the
precise
position
of
original
endoproteolytic
cleavage
was
reached
(Table
1),
i.e.
only
after
93,
158
and
223
residues
were
removed.
Other
GPI-anchored
proteins
for
which
the
cDNA
sequence
and
the
precise
C-terminal
amino
acid
of
the
mature
protein
(GPI-anchor-attachment
site)
were
available,
but
for
which
the
amino
acid
composition
was
not
reported,
were
also
analysed.
cDNA
with
the
replacement
of
Gly-SI
with
serine
[25-27],
as
We
studied
DAF
-[31,32],
in
which
the
shoTter
cDNA
sequence

Prediction
of
locus
of
endoproteolytic
cleavage
11
Table
1
Predictive
value
of
X2
statistic
comparison
among
several
GPI-anchored
proteins
using
data
from
the
cDNA-deduced
amino
acid
sequence
and
amino
acid
analysis
of
each
protein
Data
in
parentheses
indicate
the
number
of
C-terminal
amino
acids
removed
as
determined
experimentally.
*Theoretical
endoproteolytic
cleavages
(please
see
the
text
for
details).
No.
of
residues
in:
No.
of
residues
removed
No
of
excess
amino
Nascent
Mature
to
achieve
minimum
acids
removed
GPI-anchored
protein
polypeptide
protein
xI
value
(margin
of
error)
5'-Nucleotidase
(rat
liver)
DAF
(human)
CEA
(human)
PLAP
(human)
Thy-i
(rat
brain)
VSG-117
(T.
brucel)
PARP
(
T
bruce4
548
548
548
548
347
668
513
142
493
116
523
(25)
455
(93)*
385
(1
58)-
325
(223)-
319
(28)
642
(26)
484
(29)
111
(31)
470
(23)
94
(22)
25
(93)*
(158)*
(223)*
28
26
29
30
23
22
Zero
Zero
Zero
Zero
Zero
Zero
Zero
-1
Zero
Zero
(nascent
polypeptide
=
347
amino
acids)
is
post-translationally
cleaved
after
Ser-319,
the
residue
to
which
the
GPI
anchor
is
attached.
Again,
as
expected,
with
progressive
exoproteolysis
of
the
nascent
polypeptide
from
the
C-terminus,
this
value
pro-
gressively
declined
to
a
unique
zero
after
28
residues
were
removed,
leaving
Ser-319
as
the
C-terminal
amino
acid;
this
is
precisely
the
position
at
which
GPI
anchor
is
attached
to
mature
DAF
132].
A
similar
analysis
with
CEA
[28-30]
(nascent
poly-
peptide
=
668
amino
acids;
C-terminal
26
amino
acid
residues
are
post-translationally
cleaved;
GPI
anchor
attached
to
Ala-
642)
identified
that
removal
of
26
C-terminal
residues
led
to
a
unique
minimum
in
the
x2
statistic
comparison;
again,
this
method
correctly
predicted
that
Ala-642
was
indeed
the
C-
terminal
amino
acid
in
the
mature
protein
(Table
1).
Taken
together,
these
results
revealed
that
the
initial
magnitude
of
the
x2
statistic
was
reflective
of
a
difference,
albeit
minor
at
times,
between
the
mature
protein
and
nascent
polypeptide,
and
this
validated
the
assumption
that
the
mathematical
relationship
between
the
proportion
of
amino
acids
in
a
given
protein
and
its
original
sequence
could
be
objectively
defined
as
hypothesized.
In
addition,
our
method
precisely
identified
a
unique
minimum
in
the
x2
statistic
which
occurred
only
when
the
exoproteolytically
cleaved
nascent
polypeptide
had
been
reduced
in
size
to
the
'theoretically'
endoproteolytically
cleaved
mature
polypeptide
(to
which
the
GPI
anchor
was
proven
to
be
attached)
with
various-sized
GPI-anchored
proteins.
Finally,
GPI-anchored
protein
models
for
which
all
the
necessary
parameters
(cDNA
sequence,
amino
acid
composition
and
the
position
of
GPI-anchor
attachment)
had
already
been
independently
determined
were
then
subjected
to
our
statistical
approach.
The
aim
was
to
test
blindly
whether
the
method
could
objectively
predict
the
site
of
post-translational
cleavage
of
the
nascent
polypeptide
before/during
addition
of
the
preformed
GPI
anchor
by
the
putative
GPI
transamidase
[35].
In
each
of
the
following
examples,
one
of
the
authors
was
completely
unaware
of
the
known
C-terminal
amino
acid
in
the
mature
protein,
and
the
results
were
revealed
only
after
the
results
of
the
x2
analyses
were
obtained.
First,
we
focused
on
PLAP
[22-24]
(513
amino
acids;
the
locus
of
cleavage
of
the
nascent
polypeptide
and
GPI
ancho,r
attachment
previously
identified
at
Asp-484);
an
ad-
vantage
of
using
PLAP
to
validate
our
approach
lay
in
the
fact
that
it
is
tissue-specific.
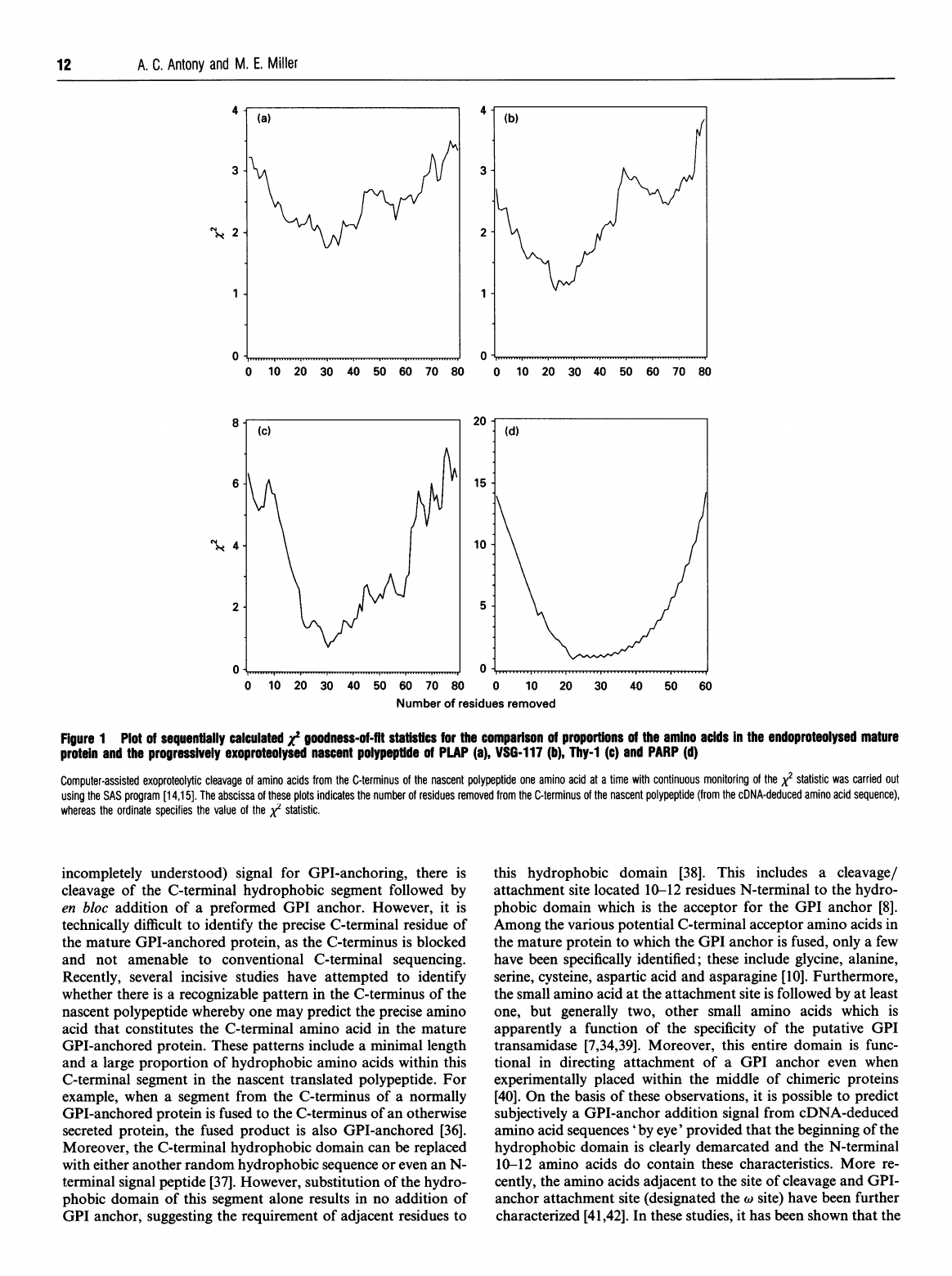
Figure
1(a)
shows
a
plot
that
'tracks'
the
x2
goodness-of-fit
statistic
calculated
when
i
residues
ar
removed
from
the
end
of
the
cDNA-deduced
PLAP
amino
acid
sequence,
and
the
resulting
proportion
of
amino
acids
are
compared
with
experimentally
generated
data
from
amino
acid
analysis
of
PLAP.
This
plot
reached
a
unique
minimum
after
29
residues
had
been
removed
from
the
nascent
polypeptide,
therebv
indicating
an
area
of
closest
agreement
between
the
exoproteolysed
(513-29
=
484
amino
acid
long)
polypeptide
and
the
mature
PLAP
polypeptide.
Of
major
significance,
this
locus
predicted
that
the
C-terminal
amino
acid
to
which
the
GPI
anchor
is
attached
in
PLAP
would
be
Asp-484,
which
is
precisely
the
residue
that
was
previously
identified
in
mature
PLAP
using
other
analytical
methods
[24].
To
verify
these
results,
we
analysed
VSG-1
17
from
T.
brucei
[20,21]
in
which
the
C-terminal
amino
acid
in
the
mature
protein
is
known
to
be
Asp-470.
The
unique
minimum
x2
value
for
this
analysis
was
1.04010
when
the
terminal
23
residues
(beginning
with
Ser-471)
were
removed
(Figure
lb).
Interestingly,
this
statistical
analysis
again
precisely
and
correctly
predicted
that
the
C-terminal
amino
acid
would
be
Asp-470.
Thus
in
both
VSG-
117
and
PLAP
there
was
no
margin
of
error
in
predicting
the
locus
of
endoproteolytic
cleavage
and
the
C-terminal
amino
acid
in
the
mature
protein.
Cys-
1I1
is
the
C-terminal
acceptor
for
the
GPI
anchor
of
Thy-
1
[18,19].
When
similarly
analysed
(Figure
Ic),
our
data
indicated
that
C-terminal
deletion
of
30
amino
acids
would
be
required
to
reach
a
point
at
which
the
proportions
of
the
C-terminal-deleted
nascent
polypeptide
and
the
mature
Thy-
l
protein
reached
maximal
agreement.
This
predicted
that
the
locus
of
endo-
proteolytic
cleavage
of
Thy-
I
nascent
polypeptide
would
be
after
amino
acid
112,
a
margin
of
error
of
one
amino
acid
from
Cys-
111
in
mature
Thy-I;
on
the
basis
of
our
studies
with
other
GPI-
anchored
proteins
(Table
1),
the
reason
for
this
discrepancy
was
probably
the
intrinsic
minor
error
associated
with
amino
acid
analysis
of
Thy-1.
PARP
cDNA
of
T.
brucei
encodes
a
GPI-anchored
protein.
Our
x2
analysis
predicted
that
the
C-terminal
22
amino
acids
would
be
removed
from
the
nascent
polypeptide
with
the
GPI
anchor
attached
to
Gly-94
of
the
mature
protein
(Figure
Id
and
Table
1);
this
was
the
precise
locus
predicted
earlier
[3]
and
also
experimentally
verified
[25-27].
iSCUSSION
The
general
rule
for
a
GPI-anchored
protein
is
that
immediately
after
translation
of
a
nascent
polypeptide
which
carries
(an
albeit
12
A.
C.
Antony
and
M.
E.
Miller
0
10
20
30
40
50
60 70
80
0
10
20
30
40
50
60
70
80
-1I
,
0....-,,,,,,,,,T,
0
10
20
30
40
50
60
70
80
0
10
Number
of
residues
removed
20
30
40
50
60
Figure
1
Plot
of
sequentially
calculated
x2
goodness-of-fit
staftstics
for
the
comparison
of
proportions
of
the
amino
acids
in
the
endoproteolysed
mature
protein
and
the
progressively
exoproteolysed
nascent
polypeptide
of
PLAP
(a),
VSG-117
(b),
Thy-i
(c)
and
PARP
(d)
Computer-assisted
exoproteolytic
cleavage
of
amino
acids
from
the
C-terminus
of
the
nascent
polypeptide
one
amino
acid
at
a
time
with
continuous
monitoring
of
the
x2
statistic
was
carried
out
using
the
SAS
program
[14,15].
The
abscissa
of
these
plots
indicates
the
number
of
residues
removed
from
the
C-terminus
of
the
nascent
polypeptide
(from
the
cDNA-deduced
amino
acid
sequence),
whereas
the
ordinate
specifies
the
value
of
the
XI
statistic.
incompletely
understood)
signal
for
GPI-anchoring,
there
is
cleavage
of
the
C-terminal
hydrophobic
segment
followed
by
en
bloc
addition
of
a
preformed
GPI
anchor.
However,
it
is
technically
difficult
to
identify
the
precise
C-terminal
residue
of
the
mature
GPI-anchored
protein,
as
the
C-terminus
is
blocked
and
not
amenable
to
conventional
C-terminal
sequencing.
Recently,
several
incisive
studies
have
attempted
to
identify
whether
there
is
a
recognizable
pattern
in
the
C-terminus
of
the
nascent
polypeptide
whereby
one
may
predict
the
precise
amino
acid
that
constitutes
the
C-terminal
amino
acid
in
the
mature
GPI-anchored
protein.
These
patterns
include
a
minimal
length
and
a
large
proportion
of
hydrophobic
amino
acids
within
this
C-terminal
segment
in
the
nascent
translated
polypeptide.
For
example,
when
a
segment
from
the
C-terminus
of
a
normally
GPI-anchored
protein
is
fused
to
the
C-terminus
of
an
otherwise
secreted
protein,
the
fused
product
is
also
GPI-anchored
[36].
Moreover,
the
C-terminal
hydrophobic
domain
can
be
replaced
with
either
another
random
hydrophobic
sequence
or
even
an
N-
terminal
signal
peptide
[37].
However,
substitution
of
the
hydro-
phobic
domain
of
this
segment
alone
results
in
no
addition
of
GPI
anchor,
suggesting
the
requirement
of
adjacent
residues
to
this
hydrophobic
domain
[38].
This
includes
a
cleavage/
attachment
site
located
10-12
residues
N-terminal
to
the
hydro-
phobic
domain
which
is
the
acceptor
for
the
GPI
anchor
[8].
Among
the
various
potential
C-terminal
acceptor
amino
acids
in
the
mature
protein
to
which
the
GPI
anchor
is
fused,
only
a
few
have
been
specifically
identified;
these
include
glycine,
alanine,
serine,
cysteine,
aspartic
acid
and
asparagine
[10].
Furthermore,
the
small
amino
acid
at
the
attachment
site
is
followed
by
at
least
one,
but
generally
two,
other
small
amino
acids
which
is
apparently
a
function
of
the
specificity
of
the
putative
GPI
transamidase
[7,34,39].
Moreover,
this
entire
domain
is
func-
tional
in
directing
attachment
of
a
GPI
anchor
even
when
experimentally
placed
within
the
middle
of
chimeric
proteins
[40].
On
the
basis
of
these
observations,
it
is
possible
to
predict
subjectively
a
GPI-anchor
addition
signal
from
cDNA-deduced
amino
acid
sequences
'by
eye'
provided
that
the
beginning
of
the
hydrophobic
domain
is
clearly
demarcated
and
the
N-terminal
10-12
amino
acids
do
contain
these
characteristics.
More
re-
cently,
the
amino
acids
adjacent
to
the
site
of
cleavage
and
GPI-
anchor
attachment
site
(designated
the
co
site)
have
been
further
characterized
[41,42].
In
these
studies,
it
has
been
shown
that
the
20
15
10
5
x4

Prediction
of
locus
of
endoproteolytic
cleavage
13
second
amino
acid
adjacent
and
C-terminal
to
the
w
site
(the
w)
+
2
site)
is
primarily
glycine
and
alanine
(and
to
a
much
lesser
extent
serine,
threonine
and
valine)
in
several
GPI-anchored
proteins.
However,
as
pointed
out
by
the
authors
of
these
elegant
studies,
the
,
co
+
2
rule
may
prove
to
be
only
75-80
%
accurate,
leading
to
the
conclusion
that
additional
determinants
for
GPI-
anchoring
also
exist
[42].
We
approached
the
issue
of
identification
of
the
locus
of
cleavage
of
the
nascent
protein
to
which
the
GPI
anchor
is
attached
using
information
from
the
cDNA
sequence
and
amino
acid
analysis
of
the
protein.
As
GPI-anchored
proteins
represent
a
truncated
form
of
the
nascent
polypeptide,
we
hypothesized
that
amino
acid
analysis
of
a
mature
GPI-anchored
protein
would
identify
a
lower
number
of
amino
acid
residues
when
compared
with
the
amino
acid
residues
from
the
cDNA-deduced
sequence.
Therefore
we
reasoned
that
a
statistical
approach
could
possibly
define
the
point
in
the
cDNA-deduced
sequence
at
which
the
nascent
polypeptide
would
probably
be
cleaved,
as
this
information
would
reside
within
the
data
from
amino
acid
analysis
of
the
mature
protein.
It
must
be
pointed
out
in
this
context
that
experimental
identification
of
the
C-terminal
amino
acid
in
GPI-anchored
proteins
is
labour-intensive,
requiring
the
isolation
of
the
GPI-anchored
protein
in
its
intact
form,
followed
by
the
generation
of
multiple
overlapping
peptides,
each
of
which
must
be
subjected
to
N-terminal
amino
acid
sequencing.
However,
determination
of
the
amino
acid
composition
of
a
purified
protein
is
a
standard,
easily
performed
and
often
routine
procedure
in
the
biochemistry
laboratory.
Therefore
we
sought
to
define
whether
use
of
the
proportion
of
amino
acids
as
obtained
from
amino
acid
analysis
would
allow
a
similar
identification
of
the
locus
of
cleavage
of
the
mature
protein.
Our
approach
involved
recalculation
of
the
x2
statistic
after
sequential
removal
of
residues
from
the
C-terminus
of
the
cDNA-
deduced
sequence
to
identify
a
point
at
which
the
x2
statistic
would
reach
a
minimum,
indicating
an
area
of
agreement
between
the
deduced
and
experimentally
verified
parameters
in
the
protein
of
interest.
The
amino
acids
that
were
deleted
would
not
be
represented
in
the
mature
protein
(which
was
subjected
to
amino
acid
analysis).
On
the
basis
of
these
theoretical
considerations,
we
validated
this
statistical
approach
by
(i)
using
theoretical
endoproteolytic
cleavages
in
the
nascent
polypeptide
and
com-
paring
the
amino
acid
proportions
of
the
product
with
that
in
the
original
nascent
polypeptide;
(ii)
performing
subsequent
studies
on
GPI-anchored
proteins
in
which
the
amino
acid
proportions
were
taken
from
the
cDNA-deduced
amino
acid
sequence
for
which
the
position
of
post-translational
cleavage
and
precise
C-
terminal
amino
acid
was
known;
and
finally,
for
(i)
and
(ii),
continuous
monitoring
of
the
x2
statistic
as
the
C-terminal
amino
acids
were
truncated
one
at
a
time
from
the
nascent
polypeptide.
In
each
of
these
instances,
the
x2
statistic
reached
a
unique
minimum
value
equal
to
zero
when
the
proportions
in
the
product
and
progressively
truncated
nascent
polypeptide
were
identical.
In
addition,
preliminary
studies
suggested
that
the
length
of
the
nascent
polypeptide
relative
to
the
length
of
the
C-
terminal
domain
cleaved
during
GPI
addition
was
not
a
limiting
factor
in
this
method
of
analysis.
These
studies
set
the
stage
for
a
comparison
between
the
proportions
of
amino
acids
from
established
GPI-anchored
species
for
which
the
data
from
amino
acid
analysis
of
the
protein
and
the
data
from
the
nascent
polypeptide
could
be
compared.
Our
experiments
on
well-studied
GPI-anchored
proteins
for
which
the
cDNA-deduced
amino
acid
sequence,
amino
acid
composition
and
C-terminal
amino
acid
were
known
confirmed
that
this
statistical
approach
could
identify
precisely
the
locus
of
cleavage
of
the
nascent
polypeptide
for
attachment
of
the
GPI
anchor.
Therefore
this
method
can
be
applied
to
other
proteins
to
identify
their
potential
for
GPI-
anchoring,
as
well
as
provide
useful
predictions
on
the
location
of
the
C-terminal
amino
acid
to
which
the
GPI
anchor
is
attached
with
a
high
degree
of
accuracy.
This
knowledge
could,
for
instance,
rapidly
help
localize
and
focus
attention
on
the
amino
acids
in
the
nascent
polypeptide
that
are
likely
targets
for
site-directed
mutagenesis
intended
to
characterize
determinants
of
membrane
insertion.
Determination
of
the
precise
amino
acid
composition
of
the
protein
is
essential
to
avoid
inaccurate
prediction
of
the
a
site.
However,
by
using
standardized
methods
and
recognizing
the
various
potential
errors
involved
in
carrying
out
an
accurate
amino
acid
analysis
of
proteins,
this
problem
can
be
circumvented
[43].
There
can
be
GPI-anchored
proteins
encoded
by
several
cDNAs
within
the
same
tissue.
One
such
species
belongs
to
the
family
of
high-affinity
folate
receptors
(folate-binding
proteins)
[44].
Human
placenta
has
been
shown
to
contain
at
least
two
distinct
placental-folate-receptor
(PFR)
cDNAs
which
are
approx.
700%
homologous
with
one
another
[45,46].
In
this
instance,
as
the
relative
proportions
and
size
of
each
PFR
isoform
in
the
purified
preparation
are
not
clear,
amino
acid
analysis
of
these
microheterogeneous
species
would
only
rep-
resent
mean
values
for
each
amino
acid
among
the
various
homologous
PFRs
[47].
Furthermore,
it
would
not
be
meaningful
to
compare
the
proportions
of
amino
acids
within
this
mixture
with
a
single
PFR
isoform
cDNA.
Thus
a
similar
analysis
of
homologous
GPI-anchored
proteins
coded
for
by
multiple
tran-
scriptionally
active
genes
is
not
likely
to
be
as
accurate
as
that
for
tissue-specific
single-gene
products.
Consequently,
it
would
be
necessary
to
overexpress
a
single
isoform
cDNA
in
a
cell
that
does
not
constitutively
express
the
protein
under
study;
sub-
sequent
isolation
and
amino
acid
analysis
of
the
GPI-anchored
protein
from
the
transfected/transduced
cell
could
allow
x2
comparisons
with
the
cDNA-predicted
nascent
polypeptide
to
define
the
locus
of
endoproteolytic
cleavage
of
the
nascent
polypeptide
isoform.
Taken
together,
our
data
suggest
that
when
the
proportions
and
number
of
residues
of
the
cDNA-deduced
and
amino-acid-
analysis-derived
data
of
any
given
protein
do
not
show
any
similarity
until
the
computer-generated
exoproteolytic
C-terminal
deletion
of
a
given
number
of
amino
acids
has
been
accomplished,
one
can
gain
information
as
to
the
locus
of
endoproteolytic
cleavage
of
the
nascent
polypeptide.
If
this
locus
includes
a
putative
GPI-anchor-attachment
site
(GPI-anchor-acceptor
amino
acids)
N-terminal
to
a
hydrophobic
domain
[41,42],
these
data
allow
prediction
of
the
likelihood
that
the
protein
under
study
has
a
GPI
anchor
added
post-translationally.
Conversely,
if
the
proportions
of
amino
acids
from
the
deduced
nascent
polypeptide
match
perfectly
with
those
obtained
from
amino
acid
analysis
(i.e.
the
x2
statistic
is
initially
zero/low
and
rises
with
progressive
computer-generated
exproteolysis
of
the
nascent
polypeptide),
then
it
suggests
that
the
mature
protein
is
anchored
by
a
(full-length)
polypeptide
membrane
anchor.
Although
we
have
not
carried
out
a
specific
prospective
study,
our
data
from
theoretical
endoproteolytic
cleavage
of
5'-nucleotidase
suggest
that
our
approach
could
also
predict
whether
a
given
protein
is
post-translationally
cleaved
by
a
protease
at
its
C-terminus
(independently
of
whether
it
is
GPI-anchored).
Thus,
if
the
computer-generated
prediction
identifies
a
locus
on
the
nascent
polypeptide
at
which
there
are
no
'acceptor'
amino
acids
N-
terminal
to
a
hydrophobic
region,
then
it
could
suggest
that the
mature
protein
was
either
post-translationally
cleaved
by
a
physiologically
relevant
protease
(in
vivo)
or
during
its
purifi-
cation
(in
vitro).

14
A.
C.
Antony
and
M.
E.
Miller
This
work
was
supported
by
grant
no.
1
R01
08307
from
the
National
Institutes
of
Health
(awarded
to
A.C.A.).
We
thank
Dr.
Mark
S.
Marshall,
Dr.
David
Leibowitz
and
Dr.
Siu
Hui
for
critically
reviewing
this
manuscript,
and
Dr.
Carol
Fiol
for
helpful
discussions.
REFERENCES
1
Moores,
S.
L.,
Schaber,
M.,
Mosser,
S.
D.,
Rands,
E.,
O'Hara,
M.
B.,
Garsky,
V.
M.,
Marshall,
M.
S.,
Pompliano,
D.
L.
and
Gibbs,
J.
B.
(1991)
J.
Biol.
Chem.
266,
14603-1
4610
2
Low,
M.
G.
(1987)
Biochem.
J.
244,
1-13
3
Ferguson,
M.
A.
J.
and
Williams,
A.
F.
(1988)
Annu.
Rev.
Biochem.
57,
285-320
4
Low,
M.
G.
(1989)
Fed.
Am.
Soc.
Exp.
Biol.
J.
3,
1600-1608
5
Doering,
T.
L.,
Masterson,
W.
J.,
Hart,
G.
W.
and
Englund,
P.
I.
(1990)
J.
Biol.
Chem.
265,
611-614
6
Cross,
G.
A.
M.
(1990)
Annu.
Rev.
Cell
Biol.
6,
1-39
7
Micanovic,
R.,
Kodukula,
K.,
Gerber,
L.
D.
and
Udenfriend,
S.
(1990)
Proc.
Natl.
Acad.
Sci.
U.S.A.
87,
7939-7943
8
Moran,
P.
and
Caras,
I.
W.
(1991)
J.
Cell
Biol.
115,
329-336
9
Moran,
P.
and
Caras,
I.
W.
(1991)
J.
Cell
Biol.
115,
1595-1600
10
Micanovic,
R.,
Gerber,
L.
D.,
Berger,
J.,
Kodukula,
K.
and
Udenfriend,
S.
(1990)
Proc.
Natl.
Acad.
Sci.
U.S.A.
87,
157-161
11
Micanovic,
R.,
Kodukula,
K.,
Gerber,
L.
D.
and
Udenfriend,
S.
(1990)
Proc.
Natl.
Acad.
Sci.
U.S.A.
87,
7939-7943
12
Lanier,
L.
L.,
Cwirla,
S.,
Yu,
G.,
Testi,
R.
and
Phillips,
J.
H.
(1989)
Science
246,
1611-1613
13
Hibbs,
M.
L.,
Selveraj,
P.,
Carpen,
O.,
Springer,
T.
A.,
Kuster,
H.,
Jouvin,
M.-H.
E.
and
Kinet,
J.-P.
(1989)
Science
246,
1608-1611
14
Davitz,
M.
A.,
Low,
M.
G.
and
Nussenzweig,
V.
(1986)
J.
Exp.
Med.
163,
1150-1161
15
Hooper,
N.
M.
and
Bashir,
A.
(1991)
Biochem.
J.
280,
745-751
16
Brown,
B.
W.
Jr.
and
Hollander,
M.
(1977)
in
Statistics:
A
Biomedical
Introduction,
p.
196-201,
John
Wiley
&
Sons,
New
York
17
SAS
Institute
Inc.
(1990)
SAS
Language:
Reference,
Version
6,
pp
1042,
Cary,
NC
18
Seki,
T.,
Chang,
H.-C.,
Moriuchi,
T.,
Denome,
R.
and
Ploegh,
H.
(1985)
Science
227,
649-651
19
Tse,
A.
G.
D.,
Barclay,
A.
N.,
Watts,
A.
and
Williams,
A.
F.
(1985)
Science
230,
1003-1008
20
Allen,
G.,
Gurnett,
L.
P.
and
Cross,
G.
A.
M.
(1982)
J.
Mol.
Biol.
157,
527-546
21
Boothroyd,
J.
C.,
Paynter,
C.
A.,
Coleman,
S.
L.
and
Cross,
G.
A.
M.
(1982)
J.
Mol.
Biol.
157,
547-556
22
Ogata,
S.,
Hayashi,
Y.,
Takami,
N.
and
Ikehara,
Y.
(1988)
J.
Biol.
Chem.
263,
10489-10494
APPENDIX
1
Description
and
reasoning
for
use
of
/
statistic
Several
statistics
could
be
used
to
measure
the
overall
discrepancy
between
the
proportions
obtained
from
amino
acid
analysis
and
the
corresponding
proportions
obtained
from
cDNA
sequencing.
The
following
discussion
initially
presents
a
general
statistic
that
can
be
used
to
measure
the
discrepancy
between
these
proportions
and
then
illustrates
how
both
the
x2
statistic
and
the
mean
squared
difference
are
two
special
cases
of
this
general
measure
(please
refer
also
to
Figure
Al).
Definitions
of
the
symbols
and
notation
used
in
the
discussion
follow:
PAi
=
the
observed
proportion
of
amino
acid
i
(i
=
1
to
D,
where
D
is
the
total
number
of
different
amino
acid
types
and
serine,
glycine
and
valine
represent
three
different
types)
measured
in
protein
X
by
amino
acid
analysis;
NA
=
the
total
number
of
amino
acids
measured
in
protein
X
by
amino
acid
analysis;
p,j
=
the
pro-
portion
of
amino
acid
i
(as
determined
from
cDNA
sequencing)
in
the
nascent
polypeptide
(protein
X+Z
amino
acids)
after
computer-generated
C-terminal
exoproteolysis
of
Y
amino
acids.
Z
is
equal
to
the
C-terminal
fragment
which
is
absent
from
the
mature
protein,
and
Y
is
increased
one
residue
at
a
time
during
computer
exoproteolysis.
For
this
application,
a
general
statistic
useful
for
summarizing
23
Henthorn,
P.
S.,
Knoll,
B.
J.,
Raducha,
M.,
Rothblum,
K.
N.,
Slaughter,
C.,
Weiss,
M.
J.,
Lafferty,
M.
A.
and
Harris,
H.
(1986)
Proc.
Natl.
Acad.
Sci.
U.S.A.
83,
5597-5601
24
Micanovic,
R.,
Bailey,
C.
A.,
Brink,
L.,
Gerber,
L.,
Pan,
Y.
C.
E.,
Hulmes,
J.
D.
and
Udenfriend,
S.
(1988)
Proc.
Natl.
Acad.
Sci.
U.S.A.
85,
1398-1402
25
Clayton,
C.
E.
and
Mowatt,
M.
R.
(1989)
J.
Biol.
Chem.
264,
15088-15093
26
Roditi,
I.,
Carrington,
M. and
Turner,
M.
(1987)
Nature
(London)
325,
272-274
27
Richardson,
J.
B.,
Beecroft,
R.
P.,
Tolson,
D.
W.,
Liu,
M.
K.
and
Pearson,
T.
W.
(1988)
Mol.
Biochem.
Parasitol.
31,
203-216
28
Oikawa,
S.,
Nakazato,
H.
and
Kosaki,
G.
(1987)
Biochem.
Biophys.
Res.
Commun.
142,
511-518
29
Paxton,
R.
J.,
Mooser,
G.,
Pande,
H.,
Lee,
T.
D.
and
Shively,
J.
E.
(1987)
Proc.
Natl.
Acad.
Sci.
U.S.A.
84,
920-924
30
Hefta,
S.
A.,
Hefta,
L.
J.
F.,
Lee,
T.
D.,
Paxton,
R.
J.
and
Shively,
J.
E.
(1988)
Proc.
Natl.
Acad.
Sci.
U.S.A.
85,
4648-4652
31
Caras,
I.
W.,
Davitz,
M.
A.,
Rhee,
L.,
Weddell,
G.,
Martin,
D.
W.,
Jr.
and
Nussenzweig,
V.
(1987)
Nature
(London)
325,
545-549
32
Moran,
P.,
Raab,
H.,
Kohr,
W.
J.
and
Caras,
I.
W.
(1991)
J.
Biol.
Chem.
266,
1250-1
257
33
Misumi,
Y.,
Ogata,
S.,
Hirose,
S.
and
Ikehara,
Y.
(1990)
J.
Biol.
Chem.
265,
21
78-21
83
34
Ogata,
S.,
Hayashi,
Y.,
Misumi,
Y.
and
Ikehara,
Y.
(1990)
Biochemistry
29,
7923-7927
35
Kodukula,
K.,
Micanovic,
R.,
Gerber,
L.,
Tamburrini,
L.,
Brink,
L.
and
Udenfriend,
S.
(1991)
J.
Biol.
Chem.
266,
4464-4470
36
Caras,
I.
W.,
Weddell,
G.
N.,
Dawitz,
M.
A.,
Nussenzweig,
V.
and
Martin,
D.
W.,
Jr.
(1987)
Science
238,
1280-1283
37
Caras,
I.
W.
and
Weddell,
G.
N.
(1989)
Science
43,
1196-1198
38
Caras,
I.
W.,
Weddell,
G.
N.
and
Williams,
S. R.
(1989)
J.
Cell
Biol.
108,
1387-1397
39
Mayor,
S.,
Menon,
A.
K.
and
Cross,
G.
A.
M.
(1991)
J.
Cell
Biol.
114,
61-71
40
Caras,
I.
W.
(1991)
J.
Cell
Biol.
113,
77-85
41
Gerber,
L.,
Kodukula,
K.
and
Udenfriend,
S.
(1992)
J.
Biol.
Chem.
267,
12168-12173
42
Kodukula,
K.,
Gerber,
L.,
Anthauer,
R.
L.,
Brink,
L.
and
Udenfriend,
S.
(1993)
J.
Cell
Biol.
120,
657-664
43
Fini,
C.,
Flovidi,
A.,
Finelli,
V.
N.
and
Wittman-Liebold,
B.
(1990)
In
Laboratory
Methodology
in
Biochemistry,
pp.
43-62,
CRC
Press,
Boca
Raton,
FL
44
Antony
A. C.
(1992)
Blood
79,
2807-2820
45
Elwood,
P.
C.
(1989)
J.
Biol.
Chem.
264,
14893-14901
46
Ratnam,
M.,
Marquardt,
H.,
Duhring,
J.
L.
and
Freisheim,
J.
H.
(1989)
Biochemistry
28,
8249-8254
47
Verma,
R.
S.,
Gullapalli,
S.
and
Antony,
A.
C.
(1992)
J.
Biol.
Chem.
267,
4119-4125
the
total
discrepancy
between
results
obtained
from
amino
acid
analysis
and
cDNA
sequencing
can
be
written
as:
D
Discrepancy
=
Wi(PAi
-pC)2
where
wi
is
a
chosen
'weight'
and
the
summation
is
over
the
D
different
amino
acid
types
contained
in
the
nascent
polypeptide
after
computer
exoproteolysis
of
Y
amino
acids.
This
statistic
measures
the
total
weighted
squared
difference
between
those
proportions
obtained
from
amino
acid
analysis
and
the
cor-
responding
proportions
obtained
from
cDNA
sequencing.
Depending
on
how
one
defines
wi,
it
is
possible
to
obtain
either
the
x2
statistic
or
a
statistic
that
measures
mean
squared
difference.
The
x2
statistic
is
obtained
by
setting
w,
=
NA/PCi1
whereas
to
obtain
the
mean
squared
difference
it
is
necessary
to
make
all
weights
equal,
w,
=
D-1,
for
every
amino
acid
type.
For
the
x2
statistic,
the
squared
difference
between
the
proportions
for
each
amino
acid
type
is
expressed
relative
to
the
proportion
of
the
amino
acid
present
in
the
cDNA-deduced
sequence.
Thus
a
difference
of
0.01
relative
to
an
underlying
proportion
of
0.30

Prediction
of
locus
of
endoproteolytic
cleavage
(a
3.33
%
error)
contributes
less
to
the
total
statistic
than
this
same
difference
relative
to
an
underlying
proportion
of
0.02
(a
50%
error).
In
contrast,
the
mean
squared
difference
statistic
'weights'
all
differences
equally
by
the
inverse
of
the
total
number
of
amino
acid
types.
The
relative
merits
of
these
two
approaches
lie
in
whether
one
believes
the
overall
statistic
should
equally 'weight'
all
magnitudes
of
random
error
that
occur
in
the
process
of
carrying
out
an
amino
acid
analysis.
Even
though
we
have
chosen
the
x2
statistic
as
our
measure
of
distance,
it
should
be
noted
that,
except
for
placental
alkaline
phosphatase,
ap-
plication
of
mean
squared
difference
to
each
of
the
examples
where
all
three
parameters
were
known
resulted
in
the
same
margin
of
error
as
reported
in
Table
1
of
the
main
paper.
For
placental
alkaline
phosphatase,
the
mean
squared
difference
did
not
identify
a
unique
minimum:
approximately
equal
statistics
were
achieved
after
dropping
29
and
34
residues.
(a)
Mature
protein
X
*
-----------------------------------
Nascent
polypeptide
(minus
N-terminal
signal
peptide)
(protein
X
+
20
amino
acids)
*----------------------------------
A
-*---------------
-
<)
(b)
Examples
1.
Protein
X
versus
protein
X
+
20
amino
acids
*->
*-------------------------------------
]0
2.
Protein
X
versus
exoproteolysis
of
protein
X
+
20
minus
10
amino
acids
*----------------------------
*---------------------------------
3.
Protein
X
versus
exoproteolysis
of
protein
X
+
20
minus
20
amino
acids
*----------------------------
*----------------------------
4.
Protein
X
versus
exoproteolysis
of
protein
X
+
20
minus
30
amino
acids
*->
Increasingly
higher
Figure
Al
Illustration
of
the
theorefical
basis
for
determination
of
the
locus
of
cleavage
of
the
nascent
polypepftde
using
/
comparison
of
the
proportions
of
amino
acids
in
a
GPI-anchored
protein
(protein
X)
and
its
nascent
polypeptide
minus
the
N-terminal
signal
peptide
(protein
X
+
20
amino
acids)
before
and
after
sequential
computer-generated
exoproteolysis
of
individual
amino
acids
from
Its
C-terminus
The
proportions
of
amino
acids
in
protein
X
are
obtained
from
amino
acid
analysis
of
the
protein,
and
proportions
of
amino
acids
in
the
nascent
polypeptide
(protein
X
+
20
amino
acids)
are
derived
from
the
cDNA-deduced
amino
acid
sequence
(minus
the
N-terminal
signal
peptide).
x2
comparisons
of
the
proportions
of
amino
acids
in
protein
X
and
protein
X
+
20
before
(example
1)
and
after
progressive
computer-generated
and
C-terminal
exoproteolysis
of
10
amino
acids
(example
2),
20
amino
acids
(example
3)
and
30
amino
acids
(example
4)
from
the
C-terminus
of
protein
X
+
20
are
shown
in
(b).
The
right-hand
column
indicates
the
expected
x2
value
when
comparisons
are
made
during
various
stages
of
C-terminal
exoproteolysis
of
protein
X
+
20
(-
n)
amino
acids.
The
asterisks
represent
the
N-terminal
amino
acid;
the
dashes
delineate
the
length
of
the
protein,
and
represent
the
various
amino
acids
comprising
the
mature
and
nascent
polypeptide.
The
arrowhead
(
A
)
in
(a)
identifies
the
locus
&,
the
C-terminal
amino
acid
to
which
the
GPI
anchor
is
attached.
The
lengths
of
the
mature
and
nascent
polypeptide
minus
the
C-terminal
fragment
[protein
X
+
20
(-20)
amino
acids]
are
identical
in
example
3
(b);
therefore
the
proportions
of
amino
acids
in
the
two
polypeptides
are
in
closest
agreement
at
this
stage
of
computer-generated
exoproteolysis
of
the
nascent
polypeptide,
theoretically
resulting
in
the
lowest
x2
value.
The
region
between
the
arrows
in
(a)
represents
the
C-terminal
fragment
of
the
nascent
polypeptide
which
is
cleaved
by
GPl
transamidase.
15
X2
value
Initially
high
Relatively
lower
Lowest
value

16
A.
C.
Antony
and
M.
E.
Miller
APPENDIX
2
Example
program
for
analysis
of
VSG-117
This
SAS
macro
implements
progressive
exoproteolysis
and
calculates
repeated
x2
statistics
for
VSG-1
17.
OPTIONS
NONOTES;
*
BEGINNING
OF
MACRO
USED
FOR
PROGRESSIVE
EXOPROTEOLYSIS;
%MACRO
DELEND(SE1
,AACID);
%DO
OBS=O
%TO
80;
DATA
A;
SET
&SEQI;
IF
N
'=
(493-&OBS);
PROC
FREQ
DATA=A;
TABLES
ACID/OUT=C1
NOPRINT;
DATA
G1;SET
Cl;
G
I
COUNT=COUNT;
KEEP
ACID
GICOUNT;
DATA
GOLD;MERGE
Gl
&MCID;BY
ACID;
RESIDUE=&OBS;
AAPCNT=RESIDUES/465;
SEQPCNT=G1COUNT/(493.&OBS);
IF
SEQPCNT>O
THEN
DO;
WI=465/SEQPCNT;
CHI=WI*(AAPCNT-SEQPCNT)**2;
OUTPUT;
END;
PROC
MEANS
SUM
NOPRINT;
VAR
CHI;
ID
RESIDUE;
OUTPUT
OUT=OUT1
SUM=CHISQ;
PROC
APPEND
BASE=FINAL
NEW=OUTI;
%END;
PROC
PLOT;
PLOT
CHISQ*RESIDUE;
%MEND;
*
END
OF
MACRO;
DATA
SEQ;
INPUT
ACID
$
@@;
CARDS;
AK
EA
L
EY
K
T
W
T
N
H
C
G
L
A
A
T
L
R
K
V
A
G
G
V
L
T
K
L
K S
HIS
Y
R
K
K
L
E
E
M
E
T
K
L
R
I
Y
A
L
K
G
D
G
V
G
E
0
K
S
A
E
IL
AT
T
AA
L
M
R
Q
K
A
L
T
P
E
E
A
N
L
K
T
A
L
K
A
A
G
F
A
G
E
G
AAA
VS
S
Y
L
M
T
L
G
T
L
T
T
S
G
S
A
H
C
L
S
N
E
G
G
D
G
D
GK
DQ
L
AP
K
G
C
R
H
G
T
E
A
D
F
D
A
G
A
G
P
A
E
S
E
V
A
D S
G
FA
Qv
P
GK
Q
D
G
A
N
A
G
a
A
N
M
C
A
L
F
T
H
Q
A
T
P
H
S
S
Q
GI
YI
T
GA
Q
T
K
P S
F
G
Y
G
M
L
T
I
G
T
T
D
Q
T
I
G
L
K
L
S
D
IK
GK
Q
AD
S
A
Q
K
F
W
S
S
C
H
A
A
V
K
A
A
Q
D
M
K
A
D
P
A
L
KV
DQ
T
LL
A
V
L
V
A
S
P
E
M
A
E
I
L
K
L
E
A
A
A
S
Q
Q
K
G
P
E
E
V
TI
D
LA
T
E
K
N
N
Y
F
G
T
N
N N
K
L
E
P
L
W
T
K
I
K
G
Q
N
IV
DL
A
AT
K
G
S
T
K
E
L
G
T
V
T
D
T
A
E
L
Q
K
L
L
S
Y
Y
Y
T
V
N
K
E
E
Q
K
K
T
A
E
K
I
T
K
L
E
T
E
L
A
D
Q
K
G
K
S
P
E
S
E
C
N
K
IS
E
E
P
K
C
N
E
D
K
I
C
S
W
H
K
E
V
K
A
G
E
K
H
C
K
F
N
S
T
K
A
K
E
K
G
VS
V T
0
T
T
A
G
G
T
E
A
T
T
D
K
C
K
G
K
L
E
O
T
C
KK
ES
N
CK
W
E
N
N
A
C
K
D
S
S
I
L
V
T
K
K
F
A
L
T V
V
S
A A
F
VA
LL
F
DATA
AMINO;
INPUT
@1
ACID
$
@6
ACIDNAME
$
6-18
@21
RESIDUES;
CARDS;
D
Aspartic
acid
23
N
Asparagine
17
T
Throonine
42
S
Serine
27
E
Glutamic
acid
40
0
Glutamine
22
P
Proline
13
G
Glycine
40
A
Alanine
59
V
Valine
17
C
Cysteine
12
M
Methionine
6
I
Isoleucine
13
L
Leucine
40
Y
Tyrosine
10
F
Phenylalanine
8
H
Histidine
9
K
Lysine
56
R
Arginine
6
W
Tryptophan
5
PROC
SORT
DATA=AMINO;BY
ACID;
TITLE1
'COMPARISON
FOR
VSG
117';
%OELEND(SEQ,AMINO);
Received
23
April
1993/13
August
1993;
accepted
17
August
1993
