
United States Office of Air and Radiation EPA 402-R-99-004A
Environmental Protection August 1999
Agency
UNDERSTANDING VARIATION IN
PARTITION COEFFICIENT, K
d
, VALUES
Volume I:
The K
d
Model, Methods of Measurement, and
Application of Chemical Reaction Codes
UNDERSTANDING VARIATION IN
PARTITION COEFFICIENT, K
d
, VALUES
Volume I:
The K
d
Model, Methods of Measurement, and
Application of Chemical Reaction Codes
August 1999
A Cooperative Effort By:
Office of Radiation and Indoor Air
Office of Solid Waste and Emergency Response
U.S. Environmental Protection Agency
Washington, DC 20460
Office of Environmental Restoration
U.S. Department of Energy
Washington, DC 20585
ii
NOTICE
The following two-volume report is intended solely as guidance to EPA and other
environmental professionals. This document does not constitute rulemaking by the Agency, and
cannot be relied on to create a substantive or procedural right enforceable by any party in
litigation with the United States. EPA may take action that is at variance with the information,
policies, and procedures in this document and may change them at any time without public notice.
Reference herein to any specific commercial products, process, or service by trade name,
trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement,
recommendation, or favoring by the United States Government.

iii
FOREWORD
Understanding the long-term behavior of contaminants in the subsurface is becoming
increasingly more important as the nation addresses groundwater contamination. Groundwater
contamination is a national concern as about 50 percent of the United States population receives
its drinking water from groundwater. It is the goal of the Environmental Protection Agency
(EPA) to prevent adverse effects to human health and the environment and to protect the
environmental integrity of the nation’s groundwater.
Once groundwater is contaminated, it is important to understand how the contaminant
moves in the subsurface environment. Proper understanding of the contaminant fate and transport
is necessary in order to characterize the risks associated with the contamination and to develop,
when necessary, emergency or remedial action plans. The parameter known as the partition (or
distribution) coefficient (K
d
) is one of the most important parameters used in estimating the
migration potential of contaminants present in aqueous solutions in contact with surface,
subsurface and suspended solids.
This two-volume report describes: (1) the conceptualization, measurement, and use of the
partition coefficient parameter; and (2) the geochemical aqueous solution and sorbent properties
that are most important in controlling adsorption/retardation behavior of selected contaminants.
Volume I of this document focuses on providing EPA and other environmental remediation
professionals with a reasoned and documented discussion of the major issues related to the
selection and measurement of the partition coefficient for a select group of contaminants. The
selected contaminants investigated in this two-volume document include: chromium, cadmium,
cesium, lead, plutonium, radon, strontium, thorium, tritium (
3
H), and uranium. This two-volume
report also addresses a void that has existed on this subject in both this Agency and in the user
community.
It is important to note that soil scientists and geochemists knowledgeable of sorption
processes in natural environments have long known that generic or default partition coefficient
values found in the literature can result in significant errors when used to predict the absolute
impacts of contaminant migration or site-remediation options. Accordingly, one of the major
recommendations of this report is that for site-specific calculations, partition coefficient values
measured at site-specific conditions are absolutely essential.
For those cases when the partition coefficient parameter is not or cannot be measured,
Volume II of this document: (1) provides a “thumb-nail sketch” of the key geochemical processes
affecting the sorption of the selected contaminants; (2) provides references to related key
experimental and review articles for further reading; (3) identifies the important aqueous- and
solid-phase parameters controlling the sorption of these contaminants in the subsurface
environment under oxidizing conditions; and (4) identifies, when possible, minimum and
maximum conservative partition coefficient values for each contaminant as a function of the key
geochemical processes affecting their sorption.

iv
This publication is the result of a cooperative effort between the EPA Office of Radiation
and Indoor Air, Office of Solid Waste and Emergency Response, and the Department of Energy
Office of Environmental Restoration (EM-40). In addition, this publication is produced as part of
ORIA’s long-term strategic plan to assist in the remediation of contaminated sites. It is published
and made available to assist all environmental remediation professionals in the cleanup of
groundwater sources all over the United States.
Stephen D. Page, Director
Office of Radiation and Indoor Air
v
ACKNOWLEDGMENTS
Ronald G. Wilhelm from ORIA’s Center for Remediation Technology and Tools was the
project lead and EPA Project Officer for this two-volume report. Paul Beam, Environmental
Restoration Program (EM-40), was the project lead and sponsor for the Department of Energy
(DOE). Project support was provided by both DOE/EM-40 and EPA’s Office of Remedial and
Emergency Response (OERR).
EPA/ORIA wishes to thank the following people for their assistance and technical review
comments on various drafts of this report:
Patrick V. Brady, U.S. DOE, Sandia National Laboratories
David S. Brown, U.S. EPA, National Exposure Research Laboratory
Joe Eidelberg, U.S. EPA, Region 9
Amy Gamerdinger, Washington State University
Richard Graham, U.S. EPA, Region 8
John Griggs, U.S. EPA, National Air and Radiation Environmental Laboratory
David M. Kargbo, U.S. EPA, Region 3
Ralph Ludwig, U.S. EPA, National Risk Management Research Laboratory
Irma McKnight, U.S. EPA, Office of Radiation and Indoor Air
William N. O’Steen, U.S. EPA, Region 4
David J. Reisman, U.S. EPA, National Risk Management Research Laboratory
Kyle Rogers, U.S. EPA, Region 5
Joe R. Williams, U.S. EPA, National Risk Management Research Laboratory
OSWER Regional Groundwater Forum Members
In addition, special acknowledgment goes to Carey A. Johnston from ORIA’s Center for
Remediation Technology and Tools for his contributions in the development, production, and
review of this document.
Principal authorship in production of this guide was provided by the Department of Energy’s
Pacific Northwest National Laboratory (PNNL) under the Interagency Agreement Number
DW89937220-01-03. Lynnette Downing served as the Department of Energy’s Project Officer
for this Interagency Agreement. PNNL authors involved in this project include:
Kenneth M. Krupka
Daniel I. Kaplan
Gene Whelan
R. Jeffrey Serne
Shas V. Mattigod
vi
TO COMMENT ON THIS GUIDE OR PROVIDE INFORMATION FOR FUTURE
UPDATES:
Send all comments/updates to:
U.S. Environmental Protection Agency
Office of Radiation and Indoor Air
Attention: Understanding Variation in Partition (K
d
) Values
401 M Street, SW (6602J)
Washington, DC 20460
or
vii
ABSTRACT
This two-volume report describes the conceptualization, measurement, and use of the partition (or
distribution) coefficient, K
d
, parameter, and the geochemical aqueous solution and sorbent
properties that are most important in controlling adsorption/retardation behavior of selected
contaminants. The report is provided for technical staff from EPA and other organizations who
are responsible for prioritizing site remediation and waste management decisions. Volume I
discusses the technical issues associated with the measurement of K
d
values and its use in
formulating the retardation factor, R
f
. The K
d
concept and methods for measurement of K
d
values
are discussed in detail in Volume I. Particular attention is directed at providing an understanding
of: (1) the use of K
d
values in formulating R
f
, (2) the difference between the original
thermodynamic K
d
parameter derived from ion-exchange literature and its “empiricized” use in
contaminant transport codes, and (3) the explicit and implicit assumptions underlying the use of
the K
d
parameter in contaminant transport codes. A conceptual overview of chemical reaction
models and their use in addressing technical defensibility issues associated with data from K
d
studies is presented. The capabilities of EPA’s geochemical reaction model MINTEQA2 and its
different conceptual adsorption models are also reviewed. Volume II provides a “thumb-nail
sketch” of the key geochemical processes affecting the sorption of selected inorganic
contaminants, and a summary of K
d
values given in the literature for these contaminants under
oxidizing conditions. The contaminants chosen for the first phase of this project include
chromium, cadmium, cesium, lead, plutonium, radon, strontium, thorium, tritium (
3
H), and
uranium. Important aqueous speciation, (co)precipitation/dissolution, and adsorption reactions
are discussed for each contaminant. References to related key experimental and review articles
for further reading are also listed.

viii
CONTENTS
Page
NOTICE ..................................................................ii
FOREWORD ............................................................. iii
ACKNOWLEDGMENTS .....................................................v
FUTURE UPDATES ....................................................... vi
ABSTRACT ..............................................................vii
LIST OF FIGURES .........................................................xii
LIST OF TABLES ........................................................ xiv
1.0 Introduction .......................................................... 1.1
2.0 The K
d
Model And Its Use In Contaminant Transport Modeling ................... 2.1
2.1 Introduction ........................................................ 2.1
2.2 Aqueous Geochemical Processes ........................................ 2.3
2.2.1 Aqueous Complexation .......................................... 2.3
2.2.2 Oxidation-Reduction (Redox) Chemistry ............................. 2.5
2.2.3 Sorption ...................................................... 2.8
2.2.3.1 Adsorption .............................................. 2.10
2.2.3.1.1 Ion Exchange ......................................... 2.13
2.2.3.2 Precipitation ............................................. 2.13
2.3 Sorption Models .................................................. 2.16
2.3.1 Constant Partition Coefficient (K
d
) Model ........................... 2.16
2.3.2 Parametric K
d
Model ........................................... 2.19
2.3.3 Isotherm Adsorption Models ..................................... 2.20
2.3.4 Mechanistic Adsorption Models ................................... 2.26
2.4 Effects of Unsaturated Conditions on Transport ........................... 2.27
2.5 Effects of Chemical Heterogeneity on Transport ........................... 2.33
2.5.1 Coupled Hydraulic and Chemical Heterogeneity ....................... 2.34
2.6 Diffusion ......................................................... 2.35
2.7 Subsurface Mobile Colloids ........................................... 2.37
2.7.1 Concept of 3-Phase Solute Transport ............................... 2.37
2.7.2 Sources of Groundwater Mobile Colloids ............................ 2.38
2.7.3 Case Studies of Mobile-Colloid Enhanced Transport of
ix
Metals and Radionuclides ........................................ 2.39
2.8 Anion Exclusion ................................................... 2.39
2.9 Summary ........................................................ 2.40
3.0 Methods, Issues, and Criteria for Measuring K
d
Values .......................... 3.1
3.1 Introduction ....................................................... 3.1
3.2 Methods for Determining K
d
Values ..................................... 3.2
3.2.1 Laboratory Batch Method ........................................ 3.3
3.2.2 In-situ Batch Method ............................................ 3.8
3.2.3 Laboratory Flow-Through Method .................................. 3.9
3.2.4 Field Modeling Method ......................................... 3.14
3.2.5 K
oc
Method .................................................. 3.14
3.3 Issues Regarding Measuring and Selecting K
d
Values ....................... 3.16
3.3.1 Using Simple Versus Complex Systems to Measure K
d
Values ............ 3.16
3.3.2 Field Variability ............................................... 3.18
3.3.3 The “Gravel Issue” ............................................. 3.19
3.3.4 The “Colloid Issue” ............................................ 3.21
3.3.5 Particle Concentration Effect ..................................... 3.22
3.4 Methods of Acquiring K
d
Values from the Literature for Screening Calculations ... 3.23
3.4.1 K
d
Look-Up Table Approach: Issues Regarding Selection of K
d
Values
from the Literature ............................................. 3.23
3.4.2 Parametric K
d
Approach ......................................... 3.26
3.4.3 Mechanistic Adsorption Models ................................... 3.28
3.5 Summary ........................................................ 3.28
4.0 Groundwater Calibration Assessment Based on Partition Coefficients:
Derivation and Examples ................................................ 4.1
4.1 Introduction ........................................................ 4.1
4.2 Calibration: Location, Arrival Time, and Concentration ...................... 4.1
4.3 Illustrative Calculations to Help Quantify K
d
Using Analytical Models ............ 4.4
4.3.1 Governing Equations ............................................ 4.4
4.3.2 Travel Time and the Partition Coefficient ............................. 4.7
4.3.3 Mass and the Partition Coefficient .................................. 4.8
x
4.3.4 Dispersion and the Partition Coefficient ............................. 4.10
4.4 Modeling Sensitivities to Variations in the Partition Coefficient ................ 4.11
4.4.1 Relationship Between Partition Coefficients and Risk ................... 4.11
4.4.2 Partition Coefficient as a Calibration Parameter in Transport Modeling ...... 4.12
4.5 Summary ........................................................ 4.14
5.0 Application of Chemical Reaction Codes ..................................... 5.1
5.1. Background ....................................................... 5.1
5.1.1 Definition of Chemical Reaction Modeling ............................ 5.2
5.1.2 Reviews of Chemical Reaction Models ............................... 5.3
5.1.3 Aqueous Speciation-Solubility Versus Reaction Path Codes ............... 5.4
5.1.4 Adsorption Models in Chemical Reaction Codes ........................ 5.5
5.1.5 Output from Chemical Reaction Modeling ............................ 5.7
5.1.6 Assumptions and Data Needs ...................................... 5.9
5.1.7 Symposiums on Chemical Reaction Modeling ......................... 5.10
5.2 MINTEQA2 Chemical Reaction Code .................................. 5.11
5.2.1 Background .................................................. 5.11
5.2.2 Code Availability .............................................. 5.11
5.2.3 Aqueous Speciation Submodel .................................... 5.12
5.2.3.1 Example of Modeling Study .................................. 5.13
5.2.3.2 Application to Evaluation of K
d
Values ......................... 5.15
5.2.4 Solubility Submodel ............................................ 5.16
5.2.4.1 Example of Modeling Study .................................. 5.17
5.2.4.2 Application to Evaluation of K
d
Values ......................... 5.18
5.2.5 Precipitation/Dissolution Submodel ................................ 5.18
5.2.5.1 Example of Modeling Study .................................. 5.19
5.2.5.2 Application to Evaluation of K
d
Values ......................... 5.20
5.2.6 Adsorption Submodel ........................................... 5.21
5.2.6.1 Examples of Modeling Studies ................................ 5.22
5.2.6.2 Application to Evaluation of K
d
Values ......................... 5.23
5.2.7 MINTEQA2 Databases ......................................... 5.24
5.2.7.1 Thermodynamic Database ................................... 5.24
5.2.7.1.1 Basic Equations ......................................... 5.25
5.2.7.1.2 Structure of Thermodynamic Database Files .................... 5.26
5.2.7.1.3 Database Components .................................... 5.26
5.2.7.1.4 Status Relative to Project Scope ............................. 5.27
5.2.7.1.5 Issues Related to Database Modifications ...................... 5.32
5.2.7.2 Sorption Database ......................................... 5.33
xi
5.2.7.2.1 Status Relative to Project Scope ............................ 5.33
5.2.7.2.2 Published Database Sources ................................ 5.33
5.3 Adsorption Model Options in MINTEQA2 ............................... 5.35
5.3.1 Electrostatic Versus Non-Electrostatic Models ........................ 5.36
5.3.2 Activity Partition Coefficient (K
d
) Model ............................ 5.40
5.3.3 Activity Langmuir Model ........................................ 5.42
5.3.4 Activity Freundlich Model ....................................... 5.44
5.3.5 Ion Exchange Model ........................................... 5.45
5.3.6 Diffuse Layer Model ........................................... 5.45
5.3.7 Constant Capacitance Model ..................................... 5.47
5.3.8 Triple Layer Model ............................................ 5.48
5.4 Summary ........................................................ 5.50
6.0 References ........................................................... 6.1
Appendix A - Acronyms, Abbreviations, Symbols, and Notation ......................A.1
Appendix B - Definitions ....................................................B.1
Appendix C - Standard Method Used at Pacific Northwest National Laboratory for
Measuring Laboratory Batch K
d
Values ..............................C.1

xii
LIST OF FIGURES
Page
Figure 2.1. Diffuse double layer and surface charge of a mineral surface ............... 2.11
Figure 2.2. Four types of adsorption isotherm curves shown schematically in
parlance of Giles et al. (1973) ...................................... 2.22
Figure 2.3. Schematic diagram for conceptual model of water distribution in
saturated (top two figures) and unsaturated soils (bottom two figures)
suggesting differences in the unsaturated flow regime (indicated by arrows)
for soils with varying texture ....................................... 2.30
Figure 2.4. Development of hydraulic heterogeneity (decreasing N
m
) in unsaturated,
non-aggregated soils with decreasing moisture saturation. ................ 2.32
Figure 3.1. Procedure for measuring a batch K
d
value (EPA 1991) .................... 3.4
Figure 3.2. Demonstration calculation showing affect on overall K
d
by multiple
species that have different individual K
d
values and are kinetically slow at
interconverting between each composition state. ........................ 3.7
Figure 3.3. Procedure for measuring a column K
d
value ............................ 3.9
Figure 3.4. Schematic diagram showing the relative concentrations of a constituent at
the input source (figures on left) and in the effluent (figures on right) as
a function of time for a pulse versus step input ......................... 3.11
Figure 4.1. Relative relationships between input-data quality, output uncertainty, and
types of problems addressed by each level of assessment ................... 4.2
Figure 4.2. Example illustrating a MEPAS
90
Sr calibration with K
d
equaling 0.8 ml/g
and 1 monitored-data point ........................................ 4.13
Figure 4.3. Example illustrating MEPAS
90
Sr calibrations with K
d
equaling 0.4
and 0.8 ml/g and several monitored-data points ......................... 4.14
Figure 5.1. Distribution of dominant U(VI) aqueous species for leachates buffered at
pH 7.0 by local ground water (Figure 5.1a) and at pH 12.5 by cement
pore fluids (Figure 5.1b) .......................................... 5.14
Figure 5.2. Saturation Indices calculated for rutherfordine (UO
2
CO
3
) as a function of
xiii
pH for solution analyses from Sergeyeva et al. (1972) .................... 5.17
Figure 5.3. Maximum concentration limits calculated for total dissolved uranium as a
function of pH based on the equilibrium solubilities of schoepite
and uranophane ................................................. 5.20
Figure 5.4. Schematic representation of the triple layer model showing surface species
and surface charge-potential relationships ............................. 5.37
Figure 5.5. Schematic representation of the constant capacitance layer model showing
surface species and surface charge-potential relationships ................. 5.38

xiv
LIST OF TABLES
Page
Table 2.1. List of several redox-sensitive metals and their possible valence states
in soil/groundwater systems ......................................... 2.6
Table 2.2. Sequence of Principal Electron Acceptors in neutral pH aquatic systems
(Sposito 1989) ................................................... 2.7
Table 2.3. Zero-point-of-charge, pH
zpc
.. ........................................ 2.9
Table 2.4. Cation exchange capacities (CEC) for several clay minerals (Grim 1968) ...... 2.14
Table 2.5. Summary of chemical processes affecting attenuation and mobility
of contaminants ................................................. 2.41
Table 3.1. Representative chemical species in acidic and basic soil solutions
(after Sposito 1989). . . ........................................... 3.17
Table 3.2. Example of a K
d
look-up table for uranium, uranium(VI), and
uranium(IV) .................................................... 3.25
Table 3.3. Advantages, disadvantages, and assumptions of K
d
determination
methods and the assumptions in applying these K
d
values to contaminant
transport models ................................................. 3.30
Table 5.1. Chemical reaction models described in the literature ....................... 5.4
Table 5.2. Examples of technical symposiums held on development, applications,
and data needs for chemical reaction modeling .......................... 5.10
Table 5.3. Component species in MINTEQA2 thermodynamic database ............... 5.29
Table 5.4. Organic ligands in MINTEQA2 thermodynamic database .................. 5.31

1
Throughout this report, the term “partition coefficient” will be used to refer to the K
d
“linear
isotherm” sorption model. It should be noted, however, that the terms “partition coefficient” and
“distribution coefficient” are used interchangeably in the literature for the K
d
model.
2
A list of acronyms, abbreviations, symbols, and notation is given in Appendix A. A list of
definitions is given in Appendix B
3
The terms “sediment” and “soil” have particular meanings depending on one’s technical
discipline. For example, the term “sediment” is often reserved for transported and deposited
particles derived from soil, rocks, or biological material. “Soil” is sometimes limited to referring
to the top layer of the earth’s surface, suitable for plant life. In this report, the term “soil” was
selected as a general term to refer to all unconsolidated geologic materials.
1.1
1.0 Introduction
The objective of this two volume report is to provide a reasoned and documented discussion on
the technical issues associated with the measurement of partition (or distribution) coefficient,
K
d
,
1,2
values and their use in formulating the contaminant retardation factor, R
f
. Specifically, it
describes the rate of contaminant transport relative to that of groundwater. The retardation factor
is the empirical parameter commonly used in transport models to describe the chemical interaction
between the contaminant and geological materials (i.e., soils, sediments, and rocks). Throughout
this report, the term “soil” will be used as general term to refer to all unconsolidated geologic
materials.
3
The contaminant retardation factor includes processes such as surface adsorption,
absorption into the soil structure, precipitation, and physical filtration of colloids. This report is
provided for technical staff from EPA and other organizations who are responsible for prioritizing
site remediation and waste management decisions.
Volume I contains a detailed discussion of the K
d
concept, its use in fate and transport computer
codes, and the methods for the measurement of K
d
values. The focus of Chapter 2 is on providing
an understanding of (1) the use of K
d
values in formulating R
f
, (2) the difference between the
original thermodynamic K
d
parameter derived from the ion-exchange literature and its
“empiricized” use in contaminant transport codes, and (3) the explicit and implicit assumptions
underlying the use of the K
d
parameter in contaminant transport codes.
The K
d
parameter is very important in estimating the potential for the adsorption of dissolved
contaminants in contact with soil. As typically used in fate and contaminant transport
calculations, the K
d
is defined as the ratio of the contaminant concentration associated with the
solid to the contaminant concentration in the surrounding aqueous solution when the system is at
equilibrium. Soil and geochemists knowledgeable of sorption processes in natural environments
have long known that generic or default K
d
values can result in significant error when used to
predict the absolute impacts of contaminant migration or site-remediation options. Therefore, for
site-specific calculations, K
d
values measured at site-specific conditions are absolutely essential.
1.2
To address some of this concern when using generic or default K
d
values for screening
calculations, modelers often incorporate a degree of conservatism into their calculations by
selecting limiting or bounding conservative K
d
values. For example, the most conservative
estimate from an off-site risk perspective of contaminant migration through the subsurface natural
soil is to assume that the soil has little or no ability to slow (retard) contaminant movement (i.e., a
minimum bounding K
d
value). Consequently, the contaminant would migrate in the direction and,
for a K
d
value of .0, travel at the rate of water. Such an assumption may in fact be appropriate
for certain contaminants such as tritium, but may be too conservative for other contaminants, such
as thorium or plutonium, which react strongly with soils and may migrate 10
2
to 10
6
times more
slowly than the water. On the other hand, to estimate the maximum risks (and costs) associated
with on-site remediation options, the bounding K
d
value for a contaminant will be a maximum
value (i.e., maximize retardation).
The K
d
value is usually a measured parameter that is obtained from laboratory experiments.
The general methods used to measure K
d
values (Chapters 3 and 4) include the laboratory batch
method, in-situ batch method, laboratory flow-through (or column) method, field modeling
method, and K
oc
method. The ancillary information needed regarding the adsorbent (soil),
solution (contaminated ground-water or process waste water), contaminant (concentration,
valence state, speciation distribution), and laboratory details (spike addition methodology, phase
separation techniques, contact times) are summarized. The advantages, disadvantages, and,
perhaps more importantly, the underlying assumptions of each method are also presented.
A conceptual overview of geochemical modeling calculations and computer codes as they pertain
to evaluating K
d
values and modeling of adsorption processes is discussed in detail in Chapter 5.
The use of geochemical codes in evaluating aqueous speciation, solubility, and adsorption
processes associated with contaminant fate studies is reviewed. This approach is compared to the
traditional calculations that rely on the constant K
d
construct. The use of geochemical modeling
to address quality assurance and technical defensibility issues concerning available K
d
data and the
measurement of K
d
values is also discussed. The geochemical modeling review includes a brief
description of the EPA’s MINTEQA2 geochemical code and a summary of the types of
conceptual models it contains to quantify adsorption reactions. The status of radionuclide
thermodynamic and contaminant adsorption model databases for the MINTEQA2 code is also
reviewed.
The main focus of Volume II is to: (1) provide a “thumb-nail sketch” of the key geochemical
processes affecting the sorption of a selected set of contaminants; (2) provide references to
related key experimental and review articles for further reading; (3) identify the important
aqueous- and solid-phase parameters controlling the sorption of these contaminants in the
subsurface environment under oxidizing conditions; and (4) identify, when possible, minimum and
maximum conservative K
d
values for each contaminant as a function key geochemical processes
affecting their sorption. The contaminants chosen for the first phase of this project include
chromium, cadmium, cesium, lead, plutonium, radon, strontium, thorium, tritium (
3
H), and
uranium. The selection of these contaminants by EPA and PNNL project staff was based on two
1.3
criteria. First, the contaminant had to be of high priority to the site remediation or risk assessment
activities of EPA. Second, due to budgetary constraints, a subset of the large number of
contaminants that met the first criteria were selected to represent categories of contaminants
based on their chemical behavior. The six nonexclusive categories are:
C Cations - cadmium, cesium, lead, plutonium, strontium, thorium, and uranium
C Anions - chromium(VI) (as chromate)
C Radionuclides - cesium, plutonium, radon, strontium, thorium, tritium (
3
H), and uranium
C Conservatively transported contaminants - tritium (
3
H) and radon
C Nonconservatively transported contaminants - other than tritium (
3
H) and radon
C Redox sensitive elements - chromium, lead, plutonium, and uranium
The general principles of geochemistry discussed in both volumes of this report can be used to
estimate the geochemical interactions of similar elements for which data are not available. For
example, contaminants present primarily in anionic form, such as Cr(VI), tend to adsorb to a
limited extent to soils. Thus, one might generalize that other anions, such as nitrate, chloride, and
U(VI)-anionic complexes, would also adsorb to a limited extent. Literature on the adsorption of
these 3 solutes show no or very little adsorption.
The concentration of contaminants in groundwater is controlled primarily by the amount of
contaminant present at the source; rate of release from the source; hydrologic factors such as
dispersion, advection, and dilution; and a number of geochemical processes including aqueous
geochemical processes, adsorption/desorption, precipitation, and diffusion. To accurately predict
contaminant transport through the subsurface, it is essential that the important geochemical
processes affecting contaminant transport be identified and, perhaps more importantly, accurately
described in a mathematically defensible manner. Dissolution/precipitation and
adsorption/desorption are usually the most important processes affecting contaminant interaction
with soils. Dissolution/precipitation is more likely to be the key process where chemical
nonequilibium exists, such as at a point source, an area where high contaminant concentrations
exist, or where steep pH or oxidation-reduction (redox) gradients exist. Adsorption/desorption
will likely be the key process controlling inorganic contaminant migration in areas where the
naturally-present constituents are already in equilibrium and only the anthropogenic constituents
(contaminants) are out of equilibrium, such as in areas far from the point source. Diffusion flux
spreads solute via a concentration gradient (i.e., Fick’s law). Diffusion is a dominant transport
mechanism when advection is insignificant, and is usually a negligible transport mechanism when
water is being advected in response to various forces.

1
For information regarding the background concentration levels of macro and trace
constituents, including elements of regulatory-interest such as arsenic, cadmium, chromium, lead,
and mercury, in soils and groundwater systems, the reader is referred to Lindsay (1979), Hem
(1985), Sposito (1989, 1994), Langmuir (1997), and other similar sources and the references
cited therein.
2
A list of acronyms, abbreviations, symbols, and notation is given in Appendix A. A list of
definitions is given in Appendix B.
2.1
2.0 The K
d
Model And Its Use In Contaminant Transport Modeling
2.1 Introduction
The concentration of contaminants in groundwater
1
is determined by the amount, concentration,
and nature of contaminant present at the source, rate of release from the source, and a number of
geochemical processes including aqueous and sorption geochemical processes (Section 2.2) and -
diffusion (Section 2.6). Recently, attention has been directed at additional geochemical processes
that can enhance the transport of certain contaminants: colloid-facilitated transport of contam-
inants (Section 2.7) and anion exclusion (Section 2.8). These latter processes are difficult to
quantify, and the extent to which they occur has not been determined. To predict contaminant
transport through the subsurface accurately, it is essential that the important geochemical
processes affecting the contaminant transport be identified and, perhaps more importantly,
accurately described in a mathematically defensible manner. Dissolution/precipitation and
adsorption/desorption are considered the most important processes affecting contaminant
interaction with soils. Dissolution/precipitation is more likely to be the key process where
chemical nonequilibium exists, such as at a waste disposal facility (i.e., point source), an area
where high contaminant concentrations exist, or where steep pH or oxidation-reduction (redox)
gradients exist. Adsorption/desorption will likely be the key process controlling contaminant
migration in areas where chemical equilibrium exists, such as in areas far from the disposal
facilities or spill sites.
The simplest and most common method of estimating contaminant retardation (i.e., the inverse of
the relative transport rate of a contaminant compared to that of water) is based on partition (or
distribution) coefficient, K
d
,
2
values (Section 2.3.1). In turn, the K
d
value is a direct measure of
the partitioning of a contaminant between the solid and aqueous phases. It is an empirical metric
that attempts to account for various chemical and physical retardation mechanisms that are
influenced by a myriad of variables. Ideally, site-specific K
d
values would be available for the
range of aqueous and geological conditions in the system to be modeled.
Values for K
d
not only vary greatly between contaminants, but also vary as a function of aqueous
and solid phase chemistry (Delegard and Barney, 1983; Kaplan and Serne, 1995; Kaplan et al.,
1994c). For example, uranium K
d
values can vary over 6 orders of magnitude depending on the
composition of the aqueous and solid phase chemistry (see Volume II, Appendix J). A more

1
A “node” is the center of a computation cell within a grid used to define the area or volume
being modeled.
2.2
robust approach to describing the partitioning of contaminants between the aqueous and solid
phases is the parametric K
d
model, which varies the K
d
value according to the chemistry and
mineralogy of the system at the node
1
being modeled (Section 2.3.2). Though this approach is
more accurate, it has not been used frequently. The added complexity in solving the transport
equation with the parametric K
d
adsorption model and its empirical nature may be why this
technique has been used sparingly.
Inherent in the K
d
“linear isotherm” adsorption model is the assumption that adsorption of the
contaminant of interest is independent of its concentration in the aqueous phase. Partitioning of a
contaminant on soil can often be described using the K
d
model, but typically only for low
contaminant concentrations as would exist some distance away (far field) from the source of
contamination. It is common knowledge that contaminant adsorption on soils can deviate from
the linear relationship required by the K
d
construct. This is possible for conditions as might exist
in leachates or groundwaters near waste sources where contaminant concentrations are large
enough to affect the saturation of surface adsorption sites. Non-linear isotherm models
(Section 2.3.3) are used to describe the case where sorption relationships deviate from linearity.
Mechanistic models explicitly accommodate the dependency of K
d
values on contaminant
concentration, competing ion concentration, variable surface charge on the absorbent, and
solution species distribution. Incorporating mechanistic or semi-mechanistic adsorption concepts
into transport models is desirable because the models become more robust and, perhaps more
importantly from the standpoint of regulators and the public, scientifically defensible. However,
less attention will be directed to mechanistic adsorption models because the focus of this project is
on the K
d
model which is currently the most common method for quantifying chemical
interactions of dissolved contaminants with soils for performance assessment, risk assessment, and
remedial investigation calculations. The complexity of installing these mechanistic adsorption
models into existing computer codes used to model contaminant transport is difficult to
accomplish. Additionally, these models also require a more intense and costly data collection
effort than will likely be available to many contaminant transport modelers, license requestors, or
responsible parties. A brief description of the state of the science and references to excellent
review articles are presented (Section 2.3.4).
The purpose of this chapter is to provide a primer to modelers and site managers on the key
geochemical processes affecting contaminant transport through soils. Attention is directed at
describing how geochemical processes are accounted for in transport models by using the
partition coefficient (K
d
) to describe the partitioning of aqueous phase constituents to a solid
phase. Particular attention is directed at: (1) defining the application of the K
d
parameter,
(2) the explicit and implicit assumptions underlying its use in transport codes, and (3) the
difference between the original thermodynamic K
d
parameter derived from ion-exchange literature
and its “empiricized” use in formulating the retardation factors used in contaminant transport

1
Unless otherwise noted, all species listed in equations in this appendix refer to aqueous
species.
2.3
codes. In addition to geochemical processes, related issues pertaining to the effects of
unsaturated conditions, chemical heterogeneity, diffusion, and subsurface mobile colloids on
contaminant transport are also briefly discussed. These processes and their effects on
contaminant mobility are summarized in a table at the end of this chapter.
2.2 Aqueous Geochemical Processes
Groundwater modelers are commonly provided with the total concentration of a number of
dissolved substances in and around a contaminant plume. While total concentrations of these
constituents indicate the extent of contamination, they give little insight into the forms in which
the metals are present in the plume or their mobility and bioavailability. Contaminants can occur
in a plume as soluble-free, soluble-complexed, adsorbed, organically complexed, precipitated, or
coprecipitated species (Sposito, 1989). The geochemical processes that contribute to the
formation of these species and their potential effect on contaminant transport are discussed in this
chapter.
2.2.1 Aqueous Complexation
Sposito (1989) calculated that a typical soil solution will easily contain 100 to 200 different
soluble species, many of them involving metal cations and organic ligands. A complex is said to
form whenever a molecular unit, such as an ion, acts as a central group to attract and form a close
association with other atoms or molecules. The aqueous species Th(OH)
4
"
(aq), (UO
2
)
3
(OH)
5
+
,
and HCO
3
-
are complexes with Th
4+
(thorium), UO
2
2+
(hexavalent uranium), and CO
3
2-
(carbonate),
respectively, acting as the central group. The associated ions, OH
-
or H
+
, in these complexes are
termed ligands. If 2 or more bonds are formed between a single ligand and a metal cation, the
complex is termed a chelate. The complex formed between Al
3+
and citric acid
[Al(COO)
2
COH(CH)
2
COOH]
+
, in which 2 COO
-
groups and 1 COH group of the citric acid
molecule are coordinated to Al
3+
, is an example of a chelate. If the central group and ligands in a
complex are in direct contact, the complex is called inner-sphere. If one or more water molecules
is interposed between the central group and a ligand, the complex is outer-sphere. If the ligands
in a complex are water molecules [e.g., as in Ca(H
2
O)
6
2+
], the unit is called a solvation complex or,
more frequently, a free species. Inner-sphere complexes usually are much more stable than outer-
sphere complexes, because the latter cannot easily involve ionic or covalent bonding between the
central group.
Most of the complexes likely to form in groundwater are metal-ligand complexes, which may be
either inner-sphere or outer-sphere. As an example, consider the formation of a neutral sulfate
complex with a bivalent metal cation (M
2+
) as the central group:
1

1
EDTA is ethylene diamine triacetic acid.
2.4
M
2%
% SO
2&
4
' MSO
"
4
(aq) (2.1)
K
r,T
'
{MSO
"
4
(aq)}
{M
2%
}{SO
2&
4
}
(2.2)
where the metal M can be cadmium, chromium, lead, mercury, strontium, etc. The equilibrium
(or stability) constant, K
r,T
, corresponding to Equation 2.1 is:
where quantities indicated by { } represent species activities. The equilibrium constant can
describe the distribution of a given constituent among its possible chemical forms if complex
formation and dissociation reactions are at equilibrium. The equilibrium constant is affected by a
number of factors, including the ionic strength of the aqueous phase, presence of competing
reactions, and temperature.
The most common complexing anions present in groundwater are HCO
3
-
/CO
3
2-
, Cl
-
, SO
4
2-
, and
humic substances (i.e., organic materials). Some synthetic organic ligands may also be present in
groundwater at contaminated sites. Dissolved PO
4
3-
can also be a strong inorganic complexant,
but is generally not very soluble in natural groundwaters. The relative propensity of the inorganic
ligands to form complexes with many metals is: CO
3
2-
> SO
4
2-
> PO
4
3-
> Cl
-
(Stumm and Morgan,
1981). Carbonate complexation may be equally important in carbonate systems, especially for
tetravalent metals (Kim, 1986; Rai et al., 1990). There can be a large number of dissolved, small-
chain humic substances present in groundwater and their complexation properties with metals and
radionuclides are not well understood. Complexes with humic substances are likely to be very
important in systems containing appreciable amounts of humic substances (>1 mg/l). In shallow
aquifers, organic ligands from humic materials can be present in significant concentration and
dominate the metal chemistry (Freeze and Cherry, 1979). The chelate anion, EDTA,
1
which is a
common industrial reagent, forms strong complexes with many cations, much stronger than
carbonate and humic substances (Kim, 1986). Some metal-organic ligand complexes can be fairly
stable and require low pH conditions (or high pH for some metal-organic complexes) to dissociate
the complex.
Complexation usually results in lowering the solution concentration (i.e., activity) of the central
molecule (i.e., uncomplexed free species). Possible outcomes of lowering the activity of the free
species of the metal include lowering the potential for adsorption and increasing its solubility, both
of which can enhance migration potential. On the other hand, some complexants (e.g., certain
humic acids) readily bond to soils and thus retard the migration of the complexed metals.
2.5
FeO(OH)(s) % 3H
%
% e
&
' Fe
2%
% 2H
2
O
(2.3)
0.5H
2
O ' 0.25O
2
(g) % H
%
% e
&
(2.4)
Fe
2%
% 1.5H
2
O % 0.25O
2
(g) ' FeO(OH)(s) % 2H
%
(2.5)
pE ' & log {e
&
} .
(2.6)
2.2.2 Oxidation-Reduction (Redox) Chemistry
An oxidation-reduction (redox) reaction is a chemical reaction in which electrons are transferred
completely from one species to another. The chemical species that loses electrons in this charge
transfer process is described as oxidized, and the species receiving electrons is described as
reduced. For example, in the reaction involving iron species:
the solid phase, goethite [FeO(OH) (s)], is the oxidized species, and Fe
2+
is the reduced species.
Equation 2.3 is a reduction half-reaction in which an electron in aqueous solution, denoted e
-
,
serves as one of the reactants. This species, like the proton in aqueous solution, is understood in
a formal sense to participate in charge transfer processes. The overall redox reaction in a system
must always be the combination of 2 half-reactions, an oxidation half-reaction and reduction half-
reaction, such that the species e
-
does not exist explicitly. For example, to represent the oxidation
of Fe
2+
, Equation 2.3 could be combined (or coupled) with the half-reaction involving the
oxidation of H
2
O:
Combining Equation 2.3 with Equation 2.4 results in the cancellation of the aqueous electron and
the oxidation of Fe
2+
via the reduction of O
2
(g) and subsequent precipitation of hydrous iron
oxide. This is a possible reaction describing Fe
2+
leaching from a reduced environment in the near
field to the oxidizing environment of the far field:
Equation 2.5 could represent a scenario in which Fe
2+
is leached from a reducing environment,
where it is mobile, into an oxidized environment, where Fe
3+
precipitates as the mineral goethite.
The electron activity is a useful conceptual device for describing the redox status of aqueous sys-
tems, just as the aqueous proton activity is so useful for describing the acid-base status of soils.
Similar to pH, the propensity of a system to be oxidized can be expressed by the negative
common logarithm of the free-electron activity, pE:
The range of pE in the natural environment varies between approximately 7 and 17 in the vadose
zone (Sposito, 1989). If anoxic conditions exist, say in a bog area, than the pE may get as low as
-3. The most important chemical elements affected by redox reactions in ambient groundwater
are carbon, nitrogen, oxygen, sulfur, manganese, and iron. In contaminated groundwater, this list

2.6
increases to include arsenic, cobalt, chromium, iodine, molybdenum, neptunium, plutonium,
selenium, technetium, uranium, and others. Table 2.1 lists several redox-sensitive metals and the
Table 2.1. List of several redox-sensitive metals and their possible valence states in
soil/groundwater systems.
Element Valence States Element Valence States
Americium +3, +4, +5, and +6 Neptunium +3, +4, +5, and +6
Antimony +3 and +5 Plutonium +2, +3, +4, +5, and +6
Arsenic +3 and +5 Ruthenium +2, +3, +4, +6, and +7
Chromium +2, +3, and +6 Selenium -2, +4, and +6
Copper +1 and +2 Technetium +2, +3, +4, +5, +6, and +7
Iron +2 and +3 Thallium +1 and +3
Manganese +2 and +3 Uranium +3, +4, +5, and +6
Mercury +1 and +2 Vanadium +2, +3, +4, and +5
different valence states that they may be present as in soil/groundwater systems. The speciation
of a metal in solution between its different valence states will depend on the site geochemistry,
especially with respect to pH and redox conditions. Moreover, not all of the valence states for
each metal are equally important from the standpoint of dominance in solution, adsorption
behavior, solubility, and toxicity. For those redox-sensitive elements that are part of this project’s
scope (i.e., chromium, plutonium, and uranium), these issues are discussed in detail in Volume II
of this report.
There is a well defined sequence of reduction of inorganic elements (Table 2.2). When an
oxidized system is reduced, the order that oxidized species disappear are O
2
, NO
3
-
, Mn
2+
, Fe
2+
, HS
-
, and H
2
. As the pE of the system drops below +11.0, enough electrons become available to
reduce O
2
(g) to H
2
O. Below a pE of 5, O
2
(g) is not stable in pH neutral systems. Above
pE = 5, O
2
(g) is consumed in the respiration processes of aerobic microorganisms. As the pE
decreases below 8, electrons become available to reduce NO
3
-
to NO
2
-
. As the system pE value
drops into the range of 7 to 5, electrons become plentiful enough to support the reduction of iron
and manganese in solid phases. Iron reduction does not occur until O
2
and NO
3
-
are depleted, but
manganese reduction can be initiated in the presence of NO
3
-
. In the case of iron and manganese,

2.7
decreasing pE results in solid-phase dissolution, because the stable forms of Mn(IV) and Fe(III)
are solid phases. Besides the increase in solution concentrations of iron and manganese expected
from this effect of lowered pE, a marked increase is usually observed in the aqueous phase
concentrations of metals such as cadmium, chromium, or lead, and of ligands such as H
2
PO
4
-
or
Table 2.2. Sequence of Principal Electron Acceptors in neutral pH aquatic
systems (Sposito, 1989).
Reduction Half-Reactions Range of Initial pE Values
0.5 O
2
(g) + 2 e
-
+ 2 H
+
= H
2
O 5.0 to 11.0
NO
3
-
+ 2 e
-
+ 2 H
+
= NO
2
-
+ H
2
O 3.4 to 8.5
MnO
2
(s) + 2 e
-
+ 4 H
+
= Mn
2+
+ 2 H
2
O 3.4 to 6.8
FeOOH (s) + e
-
+ 3 H
+
= Fe
2+
+ 2 H
2
O 1.7 to 5.0
SO
4
2-
+ 8 e
-
+ 9 H
+
= HS
-
+ 4 H
2
O 0 to -2.5
H
+
+ e
-
= 0.5 H
2
(g) -2.5 to -3.7
(CN
2
O)
n
= n/2 CO
2
(g) + n/2 CH
4
(g) -2.5 to -3.7
HMoO
4
-
, accompanying reduction of iron and manganese. The principal cause of this secondary
phenomenon is the desorption of metals and ligands that occurs when the adsorbents (i.e., mostly
iron and manganese oxides) to which they are bound become unstable and dissolve. Typically, the
metals released in this fashion, including iron and manganese, are soon readsorbed by solids that
are stable at low pE (e.g., clay minerals or organic matter) and become exchangeable surface
species.
These surface changes have an obvious influence on the availability (migration potential) of the
chemical elements involved, particularly phosphorus. If a contaminant was involved in this
dissolution/ exchange set of reactions, it would be expected that the contaminants would be less
strongly associated with the solid phase.
As pE becomes negative, sulfur reduction can take place. If contaminant metals and
radionuclides, such as Cr(VI), Pu (VI), or U(VI), are present in the aqueous phase at high enough
concentrations, they can react with bisulfide (HS
-
) to form metal sulfides that are quite insoluble.
Thus, anoxic conditions can diminish significantly the solubility of some redox-sensitive
contaminants.

2.8
Pu(IV) % e
&
' Pu(III) pE ' 1.7
(2.7)
Redox chemistry may also have a direct affect on contaminant chemistry. It can directly affect the
oxidation state of several contaminants, including, arsenic, cobalt, chromium, iodine,
molybdenum, neptunium, lead, plutonium, selenium, technetium, and uranium. A change in
oxidation in turn affects the potential of some contaminants to precipitate. For example, the
reduction of Pu(IV),
makes plutonium appreciably less reactive in complexation [i.e., Pu(III) stability constants are
much less than those of Pu(IV)] and sorption/partitioning reactions (Kim, 1986). The reduction
of U(VI) to U(III) or U(IV), has the opposite effect, i.e., U(III) or U(IV) form stronger
complexes and sorb more strongly to surfaces than U(VI). Reducing environments tend to make
chromium, similar to uranium, less mobile, and arsenic more mobile.
Therefore, changes in redox may increase or decrease the tendency for reconcentration of
contaminants, depending on the chemical composition of the aqueous phase and the contaminant
in question. However, if the redox status is low enough to induce sulfide formation,
reprecipitation of many metals and metal-like radionuclides can be expected. Redox-mediated
reactions are incorporated into most geochemical codes and can be modeled conceptually. The
resultant speciation distribution calculated by such a code is used to determine potential solubility
controls and adsorption potential. Many redox reactions have been found to be kinetically slow in
natural groundwater, and several elements may never reach redox equilibrium between their
various oxidation states. Thus, it is more difficult to predict with accuracy the migration potential
of redox-sensitive species.
2.2.3 Sorption
When a contaminant is associated with the solid phase, it is not known if it was adsorbed on to
the surface of a solid, absorbed into the structure of a solid, precipitated as a 3-dimensional
molecular structure on the surface of the solid, or partitioned into the organic matter (Sposito,
1989). Dissolution/ precipitation and adsorption/desorption are considered the most important
processes affecting metal and radionuclide interaction with soils and will be discussed at greater
lengths than absorption and organic matter partitioning.
· Dissolution/precipitation is more likely to be the key process where chemical
nonequilibium exists, such as at a point source, an area where high contaminant
concentrations exist, or where steep pH or redox gradients exist.
· Adsorption/desorption will likely be the key process controlling contaminant migration in
areas where chemical equilibrium exists, such as in areas far from the point source.

2.9
A generic term devoid of mechanism and used to describe the partitioning of aqueous phase
constituents to a solid phase is sorption. Sorption encompasses all of the above processes. It is
frequently quantified by the partition coefficient, K
d
, that will be discussed below (Section 2.3.1).
In many natural systems, the extent of sorption is controlled by the electrostatic surface charge of
the mineral phase. Most soils have net negative charges. These surface charges originate from
permanent and variable charges. The permanent charge results from the substitution of a lower
valence cation for a higher valence cation in the mineral structure, where as the variable charge
results from the presence of surface functional groups. Permanent charge is the dominant charge
of 2:1 clays, such as biotite and montmorillonite. Permanent charge constitutes a majority of the
charge in unweathered soils, such as exist in temperate zones in the United States, and it is not
affected by solution pH. Permanent positive charge is essentially nonexistent in natural rock and
soil systems. Variable charge is the dominant charge of aluminum, iron, and manganese oxide
solids and organic matter. Soils dominated by variable charge surfaces are primarily located in
semi-tropical regions, such as Florida, Georgia, and South Carolina, and tropical regions. The
magnitude and polarity of the net surface charge changes with a number of factors, including pH.
As the pH increases, the surface becomes increasingly more negatively charged. The pH where
the surface has a zero net charge is referred to as the pH of zero-point-of-charge, pH
zpc
(Table 2.3). At the pH of the majority of natural soils (pH 5.5 to 8.3), calcite, gibbsite, and
goethite, if present, would be expected to have some, albeit little, positive charge and therefore
some anion sorption capacity.
Table 2.3. pH of zero-point-of-charge, pH
zpc
. [After Stumm and
Morgan (1981) and Lehninger (1970)].
Material pH
zpc
Gibbsite [Al(OH)
3
] 5.0
Hematite ("-Fe
2
O
3
) 6.7
Goethite ("-FeOOH) 7.8
Silica (SiO
2
) 2
Feldspars 2 to 2.4
Kaolinite [Al
2
Si
2
O
5
(OH)
4
] 4.6
COOH 1.7 to 2.6
1
NH
3
9.0 to 10.4
1
1
These values represent the range of pK
a
values for amino acids.

1
The ionic potential is the ratio of the valence to the ionic radius of an ion.
2.10
2.2.3.1 Adsorption
Adsorption, as discussed in this report, is the net accumulation of matter at the interface between
a solid phase and an aqueous-solution phase. It differs from precipitation because it does not
include the development of a 3-dimensional molecular structure. The matter that accumulates in
2-dimensional molecular arrangements at the interface is the adsorbate. The solid surface on
which it accumulates is the adsorbent.
Adsorption on clay particle surfaces can take place via 3 mechanisms. In the first mechanism, an
inner-sphere surface complex is in direct contact with the adsorbent surface and lies within the
Stern Layer (Figure 2.1). As a rule, the relative affinity of a contaminant to sorb will increase
with its tendency to form inner-sphere surface complexes. The tendency for a cation to form an
inner-sphere complex in turn increases with increasing valence (i.e., more specifically, ionic
potential
1
) of a cation (Sposito, 1984).
The second mechanism creates an outer-sphere surface complex that has at least 1 water molecule
between the cation and the adsorbent surface. If a solvated ion (i.e., an ion with water molecules
surrounding it) does not form a complex with a charged surface functional group but instead
neutralizes surface charge only in a delocalized sense, the ion is said to be adsorbed in the diffuse-
ion swarm, and these ions lie in a region called the diffuse sublayer (Figure 2.1). The diffuse-ion
swarm and the outer-sphere surface complex mechanisms of adsorption involve exclusively ionic
bonding, whereas inner-sphere complex mechanisms are likely to involve ionic, as well as
covalent, bonding.
The mechanisms by which anions adsorb are inner-sphere surface complexation and diffuse-ion
swarm association. Outer-sphere surface complexation of anions involves coordination to a
protonated hydroxyl or amino group or to a surface metal cation (e.g., water-bridging
mechanisms) (Gu and Schulz, 1991). Almost always, the mechanism of this coordination is
hydroxyl-ligand exchange (Sposito, 1984). In general, ligand exchange is favored at pH levels
less than the zero-point-of-charge (Table 2.3). The anions CrO
4
2-
, Cl
-
, and NO
3
-
, and to lesser
extent HS
-
, SO
4
2-
, and HCO
3
-
, are considered to adsorb mainly as diffuse-ion and outer-sphere-
complex species.

2.11
Figure 2.1. Diffuse double layer and surface
charge of a mineral surface. (F
o
, F
s
,
and F
d
represent the surface charge
at the surface, Stern layer, and
diffuse layer, respectively; R
o
, R
s
,
and R
d
represent the potential at
the surface, Stern layer, and diffuse
layer, respectively.)
2.12
Cs
%
> Rb
%
> K
%
> Na
%
> Li
%
(2.8)
Ba
2%
> Sr
2%
> Ca
2%
> Mg
2%
(2.9)
Hg
2%
> Cd
2%
> Zn
2%
(2.10)
Fe
3%
> Fe
2%
> Fe
%
(2.11)
Cu
2%
> Ni
2%
> Co
2%
> Fe
2%
> Mn
2%
.
(2.12)
As noted previously, the relative affinity of an absorbent for a free-metal cation will generally
increase with the tendency of a cation to form inner-sphere surface complexes, which in turn
increases with higher ionic potential of a cation (Sposito, 1989). Based on these considerations
and laboratory observations, the relative-adsorption affinity of metals has been described as
follows (Sposito, 1989):
With respect to transition metal cations, however, ionic potential is not adequate as a single
predictor of adsorption affinity, since electron configuration plays a very important role in the
complexes of these cations. Their relative affinities tend to follow the Irving-Williams order:
The molecular basis for this ordering is discussed in Cotton and Wilkinson (1972).
Adsorption of dissolved contaminants is very dependent on pH. As noted previously in the
discussion of the pH of zero-point-of-charge, pH
zpc
(Table 2.3), the magnitude and polarity of the
net surface charge of a mineral changes with pH (Langmuir, 1997; Stumm and Morgan, 1981).
At pH
zpc
, the net charge of a surface changes from positive to negative. Mineral surfaces become
increasingly more negatively charged as pH increases. At pH < pH
ZPC
, the surface becomes
protonated, which results in a net positive charge and favors adsorption of contaminants present
as dissolved anions. Because adsorption of anions is coupled with a release of OH
-
ions, anion
adsorption is greatest at low pH and decreases with increasing pH. At pH > pH
ZPC
, acidic
dissociation of surface hydroxyl groups results in a net negative-charge which favors adsorption
of contaminants present as dissolved cations. Because adsorption of cations is coupled with a
release of H
+
ions, cation adsorption is greatest at high pH and decreases with deceasing pH. It
should be noted that some contaminants may be present as dissolved cations or anions depending
on geochemical conditions. In soil/groundwater systems containing dissolved carbonate, U(VI)
may be present as dissolved cations (e.g., UO
2
2+
) at low to near-neutral pH values or as anions
[e.g., UO
2
(CO
3
)
3
4-
] at near neutral to high pH values. The adsorption of U(VI) on iron oxide
minerals (Waite et al., 1994) is essentially 0 percent at pH values less than approximately 3,
increases rapidly to 100 percent in the pH range from 5 to 8, and rapidly decreases to 0 percent at
pH values greater than 9. This adsorption behavior for U(VI) (see Volume II) is reflected in the
K
d
values reported in the literature for U(VI) at various pH values.

2.13
CaX(s) % Sr
2%
' SrX(s) % Ca
2%
(2.13)
K
ex
'
{SrX(s)} {Ca
2%
}
{CaX(s)} {Sr
2%
}
(2.14)
K
d
'
{SrX(s)}
{Sr
2%
}
(2.15)
M
2%
% 2HS
&
' M(HS)
2
(s)
(2.16)
It should also be noted that the adsorption of contaminants to soil may be totally to partially
reversible. As the concentration of a dissolved contaminant declines in groundwater in response
to some change in geochemistry, such as pH, some of the adsorbed contaminant will be desorbed
and released to the groundwater.
2.2.3.1.1 Ion Exchange
One of the most common adsorption reactions in soils is ion exchange. In its most general
meaning, an ion-exchange reaction involves the replacement of 1 ionic species on a solid phase by
another ionic species taken from an aqueous solution in contact with the solid. As such, a
previously sorbed ion of weaker affinitiy is exchanged by the soil for an ion in aqueous solution.
Most metals in aqueous solution occur as charged ions and thus metal species adsorb primarily in
response to electrostatic attraction. In the cation-exchange reaction:
Sr
2+
replaces Ca
2+
from the exchange site, X. The equilibrium constant (K
ex
) for this exchange
reaction is defined by the equation:
There are numerous ion-exchange models and they are described by Sposito (1984) and Stumm
and Morgan (1981). The original usage of K
d
, often referred to as the thermodynamic K
d
, is a
special case of Equation 2.14. When one of the cations, such as Sr as the
90
Sr contaminant, is
present at trace concentrations, the amount of Ca on the exchange sites CaX(s) remains
essentially constant, as does Ca
2+
in solution. These two terms in Equation 2.14 can thus be
replaced by a constant and
The ranges of cation exchange capacity (CEC, in milliequivalents/100 g) exhibited by several clay
minerals are listed in Table 2.4 based on values tabulated in Grim (1968).
2.2.3.2 Precipitation
The precipitation reaction of dissolved species is a special case of the complexation reaction in
which the complex formed by 2 or more aqueous species is a solid. Precipitation is particularly
important to the behavior of heavy metals (e.g., nickel and lead) in soil/groundwater systems. As
an example, consider the formation of a sulfide precipitate with a bivalent radionuclide cation
(M
2+
):
2.14
K
r,T
'
{M(HS)
2
(s)}
{M
2%
} {HS
&
}
2
'
1
{M
2%
} {HS
&
}
2
(2.17)
K
sp,T
' {M
2%
} {HS
&
}
2
.
(2.18)
Table 2.4. Cation exchange capacities (CEC) for several clay minerals (Grim, 1968).
Mineral
CEC
(milliequivalents/100 g)
Chlorite 10 - 40
Halloysite · 2H
2
O 5 - 10
Halloysite · 4H
2
O 40 - 50
Illite 10 - 40
Kaolinite 3 - 15
Sepiolite-Attapulgite-Palygorskite 3 - 15
Smectite 80 - 150
Vermiculite 100 - 150
The equilibrium constant, K
r,T
, corresponding to Equation 2.16 is:
By convention, the activity of a pure solid phase is set equal to unity (Stumm and Morgan, 1981).
The solubility product, K
sp,T
, corresponding to dissolution form of Equation 2.16 is thus:
Precipitation of radionuclides is not likely to be a dominant reaction in far-field (i.e., a distance
away from a point source) or non-point source plumes because the contaminant concentrations
are not likely to be high enough to push the equilibrium towards the right side of Equation 2.16.
Precipitation or coprecipitation is more likely to occur in the near field as a result of high salt
concentrations in the leachate and large pH or pE gradients in the environment. Coprecipitation is
the simultaneous precipitation of a chemical element with other elements by any mechanism
(Sposito, 1984). The 3 broad types of coprecipitation are inclusion, absorption, and solid solution
formation.

1
An empirical solubility release model is a model that is mathematically similar to solubility, but
has no identified thermodynamically acceptable controlling solid.
2.15
Solubility-controlled models assume that a known solid is present or rapidly forms and controls
the solution concentration in the aqueous phase of the constituents being released. Solubility
models are thermodynamic equilibrium models and typically do not consider the time (i.e.,
kinetics) required to dissolve or completely precipitate. When identification of the likely
controlling solid is difficult or when kinetic constraints are suspected, empirical solubility
experiments are often performed to gather data that can be used to generate an empirical
solubility release model.
1
A solubility limit is not a constant value in a chemically dynamic system.
That is, the solubility limit is determined by the product of the thermodynamic activities of species
that constitute the solid (see Equation 2.18). If the system chemistry changes, especially in terms
of pH and/or redox state, then the individual species activities likely change. For example, if the
controlling solid for plutonium is the hydrous oxide Pu(OH)
4
, the solubility product, K
sp
, (as in
Equation 2.18) is the plutonium activity multiplied by the hydroxide activity taken to the fourth
power, i.e., {Pu}{OH}
4
= solubility product. The solubility product is fixed, but the value of
{Pu} and {OH} can vary. In fact, if the pH decreases 1 unit ({OH} decreases by 10), then for K
sp
to remain constant, {Pu} must increase by 10
4
, all else held constant. A true solubility model
must consider the total system and does not reduce to a fixed value for the concentration of a
constituent under all conditions. Numerous constant concentration (i.e., empirical solubility)
models are used in performance assessment activities that assume a controlling solid and fix the
chemistry of all constituents to derive a fixed value for the concentration of specific contaminants.
The value obtained is only valid for the specific conditions assumed.
When the front of a contaminant plume comes in contact with uncontaminated groundwater, the
system enters into nonequilibrium conditions. These conditions may result in the formation of
insoluble precipitates which are best modeled using the thermodynamic construct, K
sp
(i.e., the
solubility product described in Equation 2.18). Precipitation is especially common in groundwater
systems where the pH sharply increases. Additionally, soluble polymeric hydroxo solids of
metallic cations tend to form as the pH increases above 5 (Morel and Hering, 1993). At pH
values greater than 10, many transition metals and transuranic hydroxide species become
increasingly more soluble. The increase in solubility results from the formation of anionic species,
such as Fe(OH)
4
-
, UO
2
(CO
3
)
2
2-
, or UO
2
(CO
3
)
3
4-
. A demonstration calculation of the solubility of
U(VI) as a function of pH is given in Chapter 5. As the pH of the plume decreases from values
greater than 11 to ambient levels below approximately pH 8, some metal hydroxo solids, such as
NpO
2
(s) and
Fe(OH)
3
(s), may precipitate. The solubility behaviors of the contaminants included
in the first phase of this project are discussed in detail in the geochemistry background sections in
Volume II of this report.

2.16
A % C
i
' A
i
,
(2.19)
K
d
'
A
i
C
i
(2.20)
K
d
'
4
j
n' 1
{NpO
2
X(s)} % {NpO
2
Y(s)} % {NaNpO
2
(CO
3
)(s)} % {Na
3
NpO
2
(CO
3
)
2
(s)} % ...
4
j
n' 1
{NpO
%
2
} % {NpO
2
(OH)
%
2
} % {NpO
2
(OH)
"
(aq)} % {NpO
2
(CO
3
)
2&
2
} % ...
(2.21)
2.3 Sorption Models
2.3.1 Constant Partition Coefficient (K
d
) Model
The constant partition coefficient, K
d
, is a measure of sorption and is defined as the ratio of the
quantity of the adsorbate (i.e., metal or radionuclide) adsorbed per unit mass of solid to the
quantity of the adsorbate remaining in solution at equilibrium. For the reaction
the mass action expression for K
d
(typically in units of ml/g) is
where A = free or unoccupied surface adsorption sites,
C
i
= total dissolved adsorbate remaining in solution at equilibrium (µg/ml), and
A
i
= adsorbate on the solid at equilibrium (µg/g).
Describing the K
d
in terms of this simple reaction assumes that A is in great excess with respect to
C
i
and that the activity of A
i
is equal to 1. The K
d
term is valid only for a particular adsorbent and
applies only to those aqueous chemical conditions (e.g., adsorbate concentration,
solution/electrolyte matrix, temperature) in which it was measured. Also inherent in the K
d
term
are the assumptions that the system is reversible and is independent of the adsorbate concentration
in the aqueous phase.
Essentially all of the assumptions associated with the thermodynamically defined K
d
value
(Equation 2.20) are violated in the common protocols used to measure K
d
values for use in
contaminant transport codes. Typically, the K
d
for a given absorbent is determined in the
laboratory using soil from the study area and actual or simulated groundwater to which an
adsorbate is added at some trace concentration. The values of C
i
and A
i
are operationally defined
as the adsorbate concentrations measured in the fractions that passed through or were retained by,
respectively, filtration by some known filter pore size, such as 0.45-µm diameter. An important
practical limitation of the measurement of K
d
values is that the total concentration or radioactivity
of the adsorbate is measured, thereby treating the adsorbate as a single species. This assumption
is not an inherent requirement, but it is generally applied for convenience. A hypothetical example
of the species measured in a neptunium K
d
experiment are presented in Equation 2.21:

2.17
Conditional K
d
'
4
j
n' 1
A
i
4
j
n' 1
C
i
(2.22)
R
f
'
v
p
v
c
,
(2.23)
where { } indicate activity, and X and Y are 2 different mineral species. The solid phase in this
example contains 4 solid neptunium species, including the
species adsorbed to species X and Y
and the species precipitated as NaNpO
2
(CO
3
) and Na
3
NpO
2
(CO
3
)
2
. The dissolved phase in this
example contains 4 neptunium species including NpO
2
+
, NpO
2
(OH)
2
-
, NpO
2
(OH)
"
(aq), and
NpO
2
(CO
3
)
2
2-
. Using common laboratory techniques, experimentalist would not be able to
measure the concentrations of each of these dissolved and solid phases. Consequently, the
experimentalist can not distinguish between adsorbed and precipitated species. In this example,
there are more than just 1 dissolved and sorbed species, thereby violating an important assumption
underlying the K
d
value. Furthermore, many solutes have been observed to sorb more readily than
desorb from mineral or organic surfaces, a phenomena referred to as hysteresis.
In chemistry, the term conditional K
d
is often used (Jenne, 1977) to identify experimentally
derived partition coefficients that may not necessarily denote an equilibrium value or require some
of the other assumptions inherent in the more rigorous use of the K
d
term. The definition of
conditional K
d
is given in Equation 2.22:
where A
i
= sorbed species
C
i
= dissolved species.
Compared to Equation 2.20, Equation 2.22 more clearly represents the example represented by
Equation 2.21. No attempt will be made in this text to distinguish between the true
thermodynamic and the conditional K
d
.
An important limitation of the constant K
d
model is that it does not address sensitivity to changing
conditions. If the groundwater properties (e.g., pH and solution ionic strength) change, a
different K
d
value should be used in the model. This limitation will be discussed further in Section
2.2.3.2.
Chemical retardation, R
f
, is defined as,
where v
p
= velocity of the water through a control volume
v
c
= velocity of contaminant through a control volume.

2.18
R
f
' 1 %
D
b
n
e
K
d
(2.24)
MC
i
Mt
'
D
x
M
2
C
i
Mx
2
& v
x
MC
i
Mx
R
f
(i)
(2.25)
The chemical retardation term does not equal unity when the solute interacts with the soil; almost
always the retardation term is greater than 1 due to solute sorption to soils. In rare cases, the
retardation factor is actually less than 1, and such circumstances are thought to be caused by
anion exclusion (Section 2.8). To predict the effects of retardation, sorption processes must be
described in quantitative terms. The K
d
provides such a quantitative estimate. Knowledge of the
K
d
and of media bulk density and porosity for porous flow, or of media fracture surface area,
fracture opening width, and matrix diffusion attributes for fracture flow, allows calculation of the
retardation factor. For porous flow with saturated moisture conditions, the R
f
is defined as
where D
b
= porous media bulk density (mass/length
3
)
n
e
= effective porosity of the media at saturation.
For 1-dimensional advection-dispersion flow with chemical retardation, the transport equation can
be written as
where C
i
= concentration of contaminant species I in solution (mass/length
3
),
D
x
= dispersion coefficient of species I (length
2
/time),
v
x
= pore velocity of groundwater (length/time), and
R
f
(i) = retardation factor for species i.
For simplicity, radioactive decay has been omitted from Equation 2.25.
When the K
d
term is incorporated into the retardation factor, R
f
, as in Equation 2.24, the R
f
term
is also devoid of sorption mechanism, i.e., adsorption, absorption, or precipitation can not be
distinguished from one another as the mechanism by which the contaminants partitioned to the
solid phase. Furthermore, incorporating the K
d
term into the R
f
term assumes implicitly that the
reactions go to equilibrium and are reversible and that the chemical environment along the solute
flow path does not vary in either space or time (Muller et al.,1983). Although these assumptions
rarely hold true in the natural environment, single-value model parameters are generally employed,
with the justification that the approach builds conservatism into the analysis. Additionally, the
paucity of geochemical data at most sites precludes a more rigorous conceptual model (Section
2.3.3).
2.19
Log K
d
(Americium) ' 2.0 % 0.1[NaOH] & 26.8[HEDTA] % 153.4[HEDTA]
2
(2.26)
2.3.2 Parametric K
d
Model
Clearly, the greatest limitation of using K
d
values to calculate retardation terms (Equation 2.24) is
that it describes solute partitioning between the aqueous and solid phases for only 1 set of
environmental conditions. Such homogeneity does not exist in nature and therefore greatly
compromises the usefulness of the constant. For example, when the aqueous phase chemistry was
varied, americium K
d
values in a Hanford sediment ranged from 0.2 to 53 ml/g, roughly a 200-
fold range (Delegard and Barney, 1983). Additional variability in the americium K
d
values, albeit
less, were observed when slightly different Hanford sediments were used: 4.0 to 28.6 ml/g
(Delegard and Barney, 1983: Solution 1). Using similar aqueous phases but diverse soils,
Sheppard et al. (1976) measured americium K
d
values ranging from 125 to 43,500 ml/g.
Another practical conceptual model for adsorption is called the parametric K
d
model. The K
d
value in this model varies as a function of empirically derived relationships with aqueous and solid
phase independent parameters. Thus, it has the distinct advantage of being more robust and
removes the burden of determining new K
d
values for each environmental condition. Because the
value of a K
d
term is a function of a large number of variables, it is common to systematically vary
several parameters simultaneously in 1 experimental study. Factorial design strategies are most
often invoked to determine the systematic change resulting from varying the independent variables
on the dependent variables, typically the partition coefficient (Box and Behnken, 1960; Cochran
and Cox, 1957; Davies, 1954; Plackett and Burman, 1946). Statistical methods commonly used
to derive quantitative predictor equations include standard linear or nonlinear regression
(Snedecor and Cochran, 1967), stepwise regression (Hollander and Wolfe, 1973), and adaptive-
learning networks (Mucciardi et al., 1979, 1980). All these techniques have been used to develop
empirical relationships describing K
d
values in terms of other variables (Routson and Serne, 1972;
Serne et al., 1973; Routson et al.,1981; Delegard and Barney, 1983).
The empirical predictor equations commonly take the form of a nonlinear polynomial expression.
For example, after evaluating solutions consisting of several sodium salts, organic chelates, and
acids, Delegard and Barney (1983) derived with the following expression for an americium K
d
value:
Numerous salts were found to have no significant effect on americium K
d
values and therefore
were not included in the expression. Delegard and Barney (1983) also evaluated higher
exponential and logarithmic terms and determined that these terms did not improve the predictive
capabilities of the expression (i.e., the regression coefficients were not significant at P # 0.05).
It is critical that parametric K
d
equations, such as Equation 2.26, be used to calculate K
d
values
for systems only within the range of the independent variables used to create the equation. In the
case of Equation 2.26, the range of independent variables used to generate the model were
selected to simulate a plume emanating from a steel-lined concrete tank that contained strong

1
HEDTA is N-(2-hydroxyethyl) ethylenediaminetetraacetic acid.
2.20
caustic and high sodium contents. Using Equation 2.26 to generate americium K
d
values for a
plume low in pH and sodium concentrations would not be appropriate.
These types of statistical relationships are devoid of causality and therefore provide no certain
information on the mechanism by which the radionuclide partitioned to the solid phase, whether it
be by adsorption, absorption, or precipitation. For example, the statistical analyses may suggest a
very strong relationship between pH and the K
d
term, when the actual sorption process may be
controlled by iron oxide adsorption. Because pH and the surface charge of iron oxides are
covarients, a statistical relationship could be calculated, suggesting that sorption is solely caused
by pH.
The parametric K
d
model can be used in the retardation factor term (Equation 2.24) and the trans-
port equation (Equation 2.25). When used in the transport equation, the code must also keep
track of the current value of the independent variables (e.g., [NaOH] and [HEDTA]
1
for the
examples described in Equation 2.26) at each point in space and time to continually update the
concentration of the independent variables affecting the K
d
value. Thus, the code must track many
more parameters, and some numerical solving techniques (e.g., closed-form analytical solutions)
can no longer be used to perform the integration necessary to solve for contaminant
concentration. Generally, computer codes that can accommodate the parametric K
d
model use a
chemical subroutine to update the K
d
value used to determine the R
f
, when called by the main
transport code. The added complexity in solving the transport equation with the parametric K
d
sorption model and its empirical nature may be the reasons this approach has been used sparingly.
2.3.3 Isotherm Adsorption Models
Some adsorption studies are conducted in a systematic fashion to evaluate the effects of various
parameters on K
d
. The results of a suite of experiments evaluating the effect of contaminant
concentration on adsorption, while other parameters are held constant, are called an “adsorption
isotherm.” For soils, it is common knowledge that contaminant adsorption can deviate from the
linear relationship required by the K
d
construct discussed in Section 2.3.1. If it was possible to
keep increasing the amount of contaminant in solution contacting soil, all adsorption sites would
become saturated at some contaminant concentration and the linear relationship between
contaminant adsorbed to contaminant in solution would no longer hold. Isotherm models are
used to describe the case where sorption relationships deviate from linearity. For many short-
lived radionuclides, the mass present never reaches quantities large enough to start loading
surface adsorption sites to the point that the linear K
d
relationship is not accurate. However,
long-lived radionuclides and stable elements, such as RCRA-regulated metals, can be found in
leachates and groundwaters near waste sources at concentrations large enough to affect the
saturation of surface adsorption sites.

2.21
A
i
'
K
L
A
m
C
i
1 % K
L
C
i
(2.27)
A
i
'
A
m
C
i
B % C
i
(2.28)
A
i
' & B
A
i
C
i
% A
m
.
(2.29)
In situations where the amount of contaminant loaded on the available adsorption sites is large
enough to impact the linear adsorption construct, isotherm models are often invoked. Three
adsorption isotherm models used frequently are the Langmuir, Freundlich, and Dubinin-
Radushkevich models.
The Langmuir model was originally proposed to describe adsorption of gas molecules onto
homogeneous solid surfaces (crystalline materials) that exhibit one type of adsorption site
(Langmuir, 1918). Many investigators have tacitly extended the Langmuir adsorption model to
describe adsorption of solution species onto solid adsorbents including heterogeneous solids such
as soils. The Langmuir model for adsorption is
where A
i
= amount of adsorbate adsorbed per unit mass of solid
K
L
= Langmuir adsorption constant related to the energy of adsorption
A
m
= maximum adsorption capacity of the solid
C
i
= equilibrium solution concentration of the adsorbate.
Substituting 1/B for K
L
, one obtains
A plot of values for A
i
(y-axis) versus values of C
i
(x-axis) passes through the origin and is nearly
linear at low values of C
i
. As C
i
increases, A
i
should approach A
m
. Taking the reciprocal of
Equation 2.28 and multiplying both sides of the equation by A
i
·A
m
yields
Then, by plotting A
i
on the y-axis and (A
i
/C
i
) on the x-axis, one can determine the value of -B
from the slope of the best fit line and the value of A
m
from the intercept. Sposito (1984) and
Salter et al. (1981a) cite several instances where the Langmuir isotherm has successfully fit trace
adsorption by natural substrates. Further Sposito (1984) and Salter et al. (1981b) discuss
modifications of the Langmuir model to accommodate 2 distinct sites and competition of
2 adsorbates (the nuclide and the ion it replaces on the adsorbent) which further extend this
conceptual model’s usefulness on natural substrates. In the parlance of Giles et al. (1974) [also
see Sposito (1984)], the Langmuir adsorption isotherm is the L-curve. For L-curve isotherms, the
initial slope of A
i
(amount of solute adsorbed per unit mass of solid) [plotted on the y-axis] versus
C
i
(equilibrium solution concentration of the adsorbate) [plotted on the x-axis] is large, but the
slope decreases as C
i
increases. This forms the concave shaped curve shown in Figure 2.2. The
various curves depicted in Figure 2.2 are discussed in greater detail later in this section.

2.22
Figure 2.2. Four types of adsorption isotherm curves shown schematically in parlance
of Giles et al. (1974).
2.23
A
i
' K
F
C
N
i
(2.30)
log A
i
' log K
F
% N log C
i
(2.31)
A
i
' A
m
e
&K
DR
,
2
(2.32)
The Freundlich isotherm model (Freundlich, 1926) is defined as:
where A
i
= amount of adsorbate adsorbed per unit mass of solid
C
i
= equilibrium solution concentration of the adsorbate
K
F
= Freundlich adsorption constant
N = constant.
The Freundlich equation is sometimes written with the exponent in Equation 2.30 being 1/N
instead of N. The Freundlich model does not account for finite adsorption capacity at high
concentrations of solute, but when considering trace constituent adsorption, ignoring such
physical constraints is usually not critical. The Freundlich isotherm can be transformed to a linear
equation by taking the logarithms of both sides of Equation 2.30:
When log A
i
is plotted on the y-axis and log C
i
on the x-axis, the best-fit straight line has a slope
of N, and log K
F
is its intercept. When N=1, the Freundlich isotherm, represented by
Equation 2.31 reduces to a linear relationship. Because A
i
/C
i
is the ratio of the amount of solute
adsorbed to the equilibrium solution concentration (the definition of K
d
), the Freundlich K
F
is
equivalent to the value of K
d
.
Because adsorption isotherms at very low solute concentrations are often linear, either the
Freundlich isotherm with N equaling 1 or the Langmuir isotherm with K
L
·C
i
much greater than
1 fits the data. The value of N for the adsorption of many radionuclides is often significantly
different from 1, such that nonlinear isotherms are observed. In such cases, the Freundlich model
is a better predicator than the Langmuir model. Sposito (1984) shows how the Freundlich
isotherm is equivalent to the Langmuir isotherm where the parameter K
F
is log normally
distributed. Sposito (1984) also stresses that the Freundlich isotherm only applies to data
obtained at low values of C
i
(concentration of contaminant in the equilibrium solution).
A third adsorption model that has been used recently in nuclide studies is the Dubinin-
Radushkevich isotherm (Dubinin and Radushkevich, 1947). This model is applicable for the
adsorption of trace constituents. Should the adsorbent surface become saturated or the solute
exceed its solubility product, the model is inappropriate. The Dubinin-Radushkevich model is
more general than the Langmuir model, because it does not require either homogeneous
adsorption sites or constant adsorption potential. Its mathematical form is
where A
i
= observed amount of adsorbate adsorbed per unit mass
A
m
= sorption capacity of adsorbent per unit mass
2.24
ln A
i
' ln A
m
& K
DR
,
2
(2.33)
K
DR
= Dubinin-Radushkevich adsorption constant
, = RT 1n (1 + 1/C
i
)
R = gas constant
T = temperature (in Kelvin)
C
i
= equilibrium solution concentration of the adsorbate.
The Dubinin-Radushkevich equation can be transformed to
A plot of 1n A
i
(y-axis) versus ,
2
(x-axis) allows the estimation of 1n A
m
as the intercept and -K
DR
as the slope of the resultant straight line. Ames et al. (1982) successfully used this model to
describe adsorption of uranium and cesium onto basalt and its weathering products.
All 3 isotherm models can be compared against data from experiments that systematically vary the
mass of a trace constituent or radionuclide while holding all other parameters as constant as
possible. It is important to consider the total mass of the element present, including all stable and
other radioactive isotopes, when evaluating isotherms. It is incorrect to calculate isotherms based
on only one isotope if the system includes several (both stable and radioactive) for a particular
element. For convenience, isotherm experiments tend to consider only the total concentration or
radioactivity content and thus lumps all species for a given isotope.
It can be argued that all 3 isotherm models are based on physicochemical processes or
mechanisms. If the experiments are performed and characterized rigorously to assure equilibrium
conditions and constancy of variables aside from the trace constituent concentration, then the
resultant isotherm constants undoubtedly have some relationship to adsorption capacities and to
site adsorption energies. On the other hand, any suite of experiments that can be plotted as
amount adsorbed versus amount in solution at the time of measurement can also be analyzed
using these models to see whether predictive equations can be determined. The latter empirical
approach is a step up in sophistication over the constant K
d
model.
Giles et al. (1974) state that isotherm shapes are largely determined by the adsorption mechanism,
and thus can be used to explain the nature of adsorption. The S-curve (Figure 2.2) is explained by
Giles et al. (1974) as adsorption where the presence of the individual solute molecules bound to
the solid interact with each other. This increases the strength of the individual solute bonds to the
solid surface when the solid has low contaminant loading. Thus, for a brief period during
adsorption, the first bound molecules enhance adsorption of the next molecules that bind to the
solid. The slope of the isotherm increases from the lowest concentration of contaminant where
surface coverage is so sparse that adsorbed molecules cannot interact. At some point, the sites
become laden with contaminant and the slope of the adsorption isotherm starts decreasing again.
Sposito (1984) gives another plausible explanation for the S-curve, wherein complexing solution
ligands compete with the surface sites for the contaminant until the complexing ligands are
2.25
complexed with the contaminant and additional contaminant is free to adsorb with less or no
complexant competition.
The L-curve (Figure 2.2) is the classical Langmuir curve where the loading of contaminant on the
solid starts to decrease the adsorption slope as sites become saturated. The adsorption of many
RCRA-regulated metals and long-lived radionuclides on soils have been successfully described by
Langmuir isotherms. Giles et al. (1974) shows that the L-shaped curve is found for systems
where the activation energy for the adsorption/desorption of each adsorbate is unaffected by the
other adsorbates and solvent (water) in the systems. Rai and Zachara (1984) include one
compilation of Langmuir isotherms for RCRA-regulated metals.
The H-curve (Figure 2.2) is an extreme version of the L-curve isotherm. The H-curve describes
adsorption of “high-affinity” adsorbates onto solid adsorbents. The activation energy of the
desorption of the analyte of interest is much larger than other species in the solution.
The C-curve (constant slope) (Figure 2.2) suggests that the number of available sorption sites
remains constant throughout the whole range of solute concentrations (whereas the K
d
model
applies to low solute concentrations) or the available surface expands proportionally with the
amount of material adsorbed up to the point where all adsorption sites are filled. Giles et al.
(1974) discuss two conceptual models on how the available sorption sites can expand in
proportion to the adsorbed mass. The model where the adsorbent is microporous and the
adsorbate has a much higher affinity for the adsorbent surfaces than the water is most germane for
soils. The adsorbates enter the microporous solid and act like a molecular wedge to open up
more sorption sites through continued penetration.
It is difficult to assess whether one should put much weight on isotherm shape constructs
discussed by Giles et al. (1974) as a vehicle to elaborate on adsorption mechanisms. The number
of discrete crystalline minerals and amorphous phases and coatings present in soils as well as the
multitude of inorganic and organic ligands found or expected in soil solutions combine to make a
quantitative description of contaminant adsorption and the controlling mechanisms a formidable
activity. To quote Sposito (1984, pg. 122), “The adherence of experimental sorption data to an
adsorption isotherm provides no evidence as to the actual mechanism of the sorption process in
soils and sediments.” We value this statement and suggest that isotherms are just one step more
sophisticated than the constant K
d
construct in delineating or quantifying adsorption of
contaminants.
It must be stressed that isotherm models, as expressed by Equations 2.27, 2.30, and 2.32,
explicitly consider dependency of the partition coefficient on only the solution concentration of
the contaminant of interest. Isotherm models do not consider dependence on other solid and
solution parameters that can influence adsorption, such as those discussed in Volume II for each
contaminant of interest.
2.26
The incorporation of adsorption isotherm models into transport codes is relatively easy. Each of
the aforementioned isotherm equations can be rearranged to calculate a partition coefficient, K
d
,
that is a function of C, the solution concentration of the radionuclide, and 1 or 2 constants. As
the transport model solves for C, substitution of an equation that depends only upon C (and
derivable constants) for the K
d
in the retardation factor (see Equations 2.23 and 2.24) should be
straightforward. For simple cases, analytical closed-formed solutions are possible, or numerous
numerical approximation schemes can be used. Thus, with little additional work or increases in
computer storage requirements, most transport codes can be formulated to predict radionuclide
migration with an adsorption isotherm model. It should repeated, however, that this approach
accounts for the dependency of K
d
on only one parameter, the concentration of the radionuclide.
If the mass of contaminant in the environment is low, and complicating factors such as
complexing agents and type S sorption behavior are not expected, all 3 adsorption isotherms
discussed above are readily simplified to the constant K
d
model.
2.3.4 Mechanistic Adsorption Models
Mechanistic models explicitly accommodate for the dependency of K
d
values on contaminant con-
centration, competing ion concentration, variable surface charge on the adsorbent, and solute
species solution distribution. Incorporating mechanistic, or semi-mechanistic, adsorption
concepts into transport models is attempted because the models become more robust and, perhaps
more importantly from the standpoint of regulators and the public, scientifically defensible.
However, less attention will be directed to these adsorption models because we judge them of
little practical use for the majority of site-screening applications. The complexity of installing
these mechanistic adsorption models into existing transport codes is difficult to accomplish.
Additionally, mechanistic adsorption models also require a more intense and costly data collection
effort than will likely be available to the majority of EPA, DOE, and NRC contaminant transport
modelers and site remediation managers. A brief description of the state of the science is
presented below. References to excellent review articles have been included in the discussion to
provide the interested reader with additional information.
Experimental data on interactions at the mineral-electrolyte interface can be represented mathe-
matically through 2 different approaches: (1) empirical models and (2) mechanistic models. An
empirical model can be defined as a mathematical description of the experimental data without any
particular theoretical basis. For example, the K
d
, Freundlich isotherm, Langmuir isotherm,
Langmuir Two-Surface Isotherm, and Competitive Langmuir construct are considered empirical
models by this definition (Sposito, 1984). Mechanistic models refer to models based on
thermodynamic concepts such as reactions described by mass action laws and material balance
equations. Four of the most commonly used mechanistic models include the Helmholtz, Gouy-
Chapman, Stern, and Triple Layer models (Sposito, 1984). The empirical models are often
mathematically simpler than mechanistic models and are suitable for characterizing sets of
experimental data with a few adjustable parameters, or for interpolating between data points. On
the other hand, mechanistic models contribute to an understanding of the chemistry at the
interface and are often useful for describing data from complex multicomponent systems for
2.27
which the mathematical formulation (i.e., functional relationships) for an empirical model might
not be obvious. Mechanistic models can also be used for interpolation and characterization of
data sets in terms of a few adjustable parameters (Westall, 1986). However, it is important to
realize that adjustable parameters are required for both mechanistic and empirical models, except
the K
d
model. The need to include adjustable parameters in order to apply/solve mechanistic
models compromises their universal application.
Any complete mechanistic description of chemical reactions at the mineral-electrolyte interface
must include a description of the electrical double layer (Figure 2.1). While this fact has been
recognized for years, a satisfactory description of the double layer at the mineral-electrolyte
interface still does not exist. Most electrical double layer models were written for specific
conditions and are only accurate under limited environmental conditions. For instance, the Stern
model is a better model for describing adsorption of inner-sphere complexes, whereas the Gouy-
Chapman model is a better model for describing outer-sphere or diffuse swarm adsorption
(Sposito, 1984; Westall, 1986) (Figure 2.1).
Truly mechanistic models are rarely, if ever, applied to complex natural soils (Schindler and
Sposito, 1991; Sposito, 1984; Westall and Hohl, 1980, Westall, 1986; Westall, 1994). The
primary reason for this is because the surfaces of natural mineral are very irregular and difficult to
characterize. These surfaces consist of different microcrystalline structures and/or coatings of
amorphous phases that exhibit quite different and complex chemical properties when exposed to
solutions. Thus, examination of the surface by virtually any experimental method yields only
averaged characteristics of the surface and the interface. Parsons (1982) discussed the surface
chemistry of single crystals of pure metals and showed that the potential of zero charge of
different crystal faces of the same pure metal can differ by over 400 mV. For an oxide surface,
this difference was calculated by Westall (1986) to be energetically equivalent to a variation in the
pH of zero-point-of-charge (pH
zpc
) of more than 6 pH units. This example indicated that an
observable macroscopic property of a polycrystalline surface might be the result of a combination
of widely different microscopic properties and that characterizations of these surfaces will remain
somewhat operational in nature.
Another fundamental problem encountered in characterizing reactions at the mineral-electrolyte
interface is the coupling between electrostatic and chemical interactions, which makes it difficult
to distinguish the effects of one from the effects of the other. Westall and Hohl (1980) have
shown that many models for reactions at the mineral-electrolyte interface are indeterminate in this
regard.
2.4 Effects of Unsaturated Conditions on Transport
The major pathway for contaminant transport in arid areas is through unsaturated soils. Although
considerable effort has been expended over the past few years to quantify the mobility of
contaminants and determine factors that influence contaminant mobility, little work has been done
to investigate the transport of radionuclides under conditions of partial saturation.

1
The flux density is the volume of water flowing through a cross-section area per unit time.
2
The hydraulic gradient is the head drop per unit distance in the flow direction.
2.28
At unsaturated moisture conditions, the pores are partially filled with air and water. The water in
the pores is partly held in place by attractive forces of capillarity. A key hydrologic measurement
in unsaturated (vadose zone) soils is the soil-water matric potential (or suction). Matric potential
is defined as, the amount of work that must be done per unit of soil solution in order to transport,
reversibly and isothermally, an infinitesimal quantity of water from a pool of soil solution at a
given elevation above the water table (and at atmospheric pressure) to the soil pores at the same
elevation and pressure (SSSA, 1997). When the work (or energy) is expressed on a weight basis,
the matric potential is expressed in units of length (i.e., m or cm). Matric potential is always
negative (i.e., energy is gained in going from a saturated solution to unsaturated soil pores,
because of adsorptive forces and capillarity of porous material). Matric suction is the absolute
value of matric potential and is used for convenience to express the matric forces (potentials) as
positive values. By definition, at the water table, both the matric potential and matric suction are
zero.
Unsaturated flow properties include the unsaturated hydraulic conductivity and the water
retention characteristics (relationship between water content and matric suction values).
Analogous to saturated flow where the advective flux
1
is the product of the saturated hydraulic
conductivity and the gradient of the hydrostatic head,
2
the advective flow in unsaturated
sediments is the product of the unsaturated conductivity and the matric potential (or suction)
gradient. The suction gradient defines the direction of flow (from areas of low to high suction).
At most vadose zone sites there have been no direct measurements of either the unsaturated
conductivity or water retention characteristics for sediments. Generally only water contents have
been measured (often by neutron logging) in boreholes or from split-spoon samples.
When the soil is saturated, nearly all pores are filled and hydraulic conductivity is at a maximum.
As the soil becomes unsaturated, some of the pores become air-filled and the conductive cross-
sectional areas are decreased. In addition, the first pores to empty are the largest and most
conductive and tortuosity is increased for any water molecule that is still actively advecting
through the porous media (i.e., the water must find less direct pathways around these empty
pores). In unsorted soils, the large pores that resulted in high conductivity at saturation become
barriers to liquid flow between smaller pores during unsaturated flow. Hence, the transition from
saturated to unsaturated flow may result in a steep drop in hydraulic conductivity of several
orders of magnitude as the tension increase from 0 to 1 bar. At higher tensions (i.e., more
unsaturation), conductivity may be so low that steep pressure gradients are required for any
appreciable soil water flow to occur. An interesting corollary of the pore size-conductivity
relationship is that at, or near, saturation, a sandy soil conducts water more rapidly than a clay soil
with many micropores. When the soils are unsaturated, however, many of the micropores in the
clay soil remain filled, and consequently, the hydraulic conductivity in the clay soil does not
decrease nearly as sharply as it does in sandy soil under the same tension. If the soil water does

2.29
R
f
' 1 %
D
b
2
K
d
(2.34)
not move, then the contaminant in, or contacted by, the soil water does not move except by
diffusion (Section 2.6), which is a relatively slow process (Rancon, 1973).
In modeling contaminant transport in unsaturated conditions, Equation 2.24 takes the form:
where 2, the volumetric water content of the soil (cm
3
water/cm
3
total), replaces n (cm
3
void
space/cm
3
total), soil porosity, in Equation 2.24. Equation 2.27 explicitly assumes that the extent
to which contaminants sorb to soils, the K
d
value, is constant as a function of the volumetric water
content. This relationship is convenient for modeling; however, its validity is not certain. There
have been experiments to test this assumption, and the results have been mixed (Gee and
Campbell, 1980; Knoll, 1960; Lindenmeier et al., 1995; Nielsen and Biggar, 1961; Nielsen and
Biggar, 1962; Routson and Serne, 1972).
There are theoretical reasons for believing that K
d
values vary as a function of volumetric water
content. First, as the soil becomes increasingly unsaturated there will be a smaller percentage of
the total exchange sites in contact with the aqueous phase. For example, if only half of the
exchange sites of a soil come into contact with the aqueous phase, then the effective exchange
capacity of the soil is only half of that, had all the available exchange sites come into contact with
the aqueous phase. Therefore, as less mineral surface is exposed to the aqueous phase, the lower
the effective exchange capacity becomes because less of the surface is exposed to the solute of
interest. On the other hand, the clay fraction of the soil constitutes the largest exchange capacity
and smallest pore sizes. Because the smaller pores are involved in unsaturated flow, there may be
little measurable effect on the exchange capacity of the soil in unsaturated conditions. Another
reason for believing that K
d
values would vary with degree of saturation is because in the
unsaturated systems the aqueous phase is in closer contact with the soil surfaces. Solutes in the
middle of large pores have less interaction with soil surfaces than solutes nearer to the soil
surfaces. In unsaturated conditions, the middle of large pores tend to be empty, resulting in a
greater percentage of pore water being in close contact with the soil surface. Finally, the ionic
strength of the aqueous phase tends to increase closer to the clay surfaces. Thus, as a soil
dehydrates, the system tends to have a higher ionic strength. The K
d
value for many cations tends
to decrease with increases in ionic strength.
The average size of individual pores is larger for coarse- versus fine-textured soils, despite the
finer-grained soils having a larger total porosity. In saturated soil, all of the pore space is water-
filled; the pores are continuous or “connected,” and generally water conducting. As a saturated
soil is desaturated, the larger pores drain first, and air becomes a barrier to water flow. Water
flow in unsaturated soils may occur as film flow along the particle surface, or as “matrix flow”
through smaller, water-filled pores. The unsaturated flow regime is expected to differ in
unsaturated coarse- versus fine-textured soils, with film flow dominating the former, and matrix
flow the latter. This conceptualizations is illustrated in Figure 2.3. Recent improvements in

2.30
2.31
As a soil is progressively desaturated, retained water is held with increasingly greater “suction,”
expressed as the matric or negative pressure potential. Hydraulic conductivity, K
h
(m/sec) reflects
the ease of water flow through the media and decreases with decreasing moisture saturation (i.e.,
greater resistance to flow at lower water contents). Hydraulic conductivity is also highly
dependent on soil texture. Relationships between hydraulic conductivity, matric potential, and
water content are well-established (Hillel, 1998; Jury et al., 1991). The spatial variability of
hydraulic conductivity and the effect on solute transport in saturated soils have been the subject of
modeling investigations (e.g., Dagan, 1984; Gelhar and Axness, 1983; Tompson and Gelhar,
1990). Various modeling approaches are reviewed by Koltermann and Gorelick (1996) and
compared to field data by Sudicky (1986). For unsaturated soils, research has focussed on
scaling, and the effect of spatially variable hydraulic conductivity on water flow, infiltration, and
drainage (Hopmans et al.,1988; Nielsen et al.,1973; Peck et al., 1977; Warrick and Amoozegar-
Fard, 1979).
In unsaturated soils, the pathway for water flow can become more tortuous, and water held in
films and in small pores can be “disconnected” with respect to the flow regime. In reviewing
water flow and transport in the vadose zone, Nielsen et al. (1986) noted that in addition to water
held within aggregates, immobile water may exist in thin liquid films around soil particles, in dead-
end pores, or as relatively isolated regions associated with unsaturated flow. Immobile water is
also apparent in saturated systems. Most investigations that consider mass transfer between
mobile and immobile water regions have been conducted in saturated systems.
Immobile water is manifest as hydraulic heterogeneity, and is typically characterized with a “dual
porosity” or “two-region” model (Coats and Smith, 1964; Haggerty and Gorelick, 1995; van
Genuchten and Wierenga, 1976). The liquid phase is partitioned into mobile and immobile
(stagnant or micro-porosity) regions, where advective solute transport is limited to the mobile
water phase. The mobile water fraction, N
m
, is defined as the volume fraction of water associated
with the mobile domain, 2
m
, relative to the total water content, 2
v
(i.e., N
m
= 2
m
/2
v
). Transport in
and out of the immobile water domain is diffusion-limited. A small degree of hydraulic
heterogeneity can be characterized by increased hydrodynamic dispersion (Pickens et al.,1981).
However, a 2-region flow regime is required as the fraction of stagnant water increases. The
importance of particle scale properties (Ball et al., 1990), specifically mass transfer between
mobile and immobile water regions (Haggerty and Gorelick, 1995), in affecting larger-scale
transport has been noted. Compared to the standard advection-dispersion model with sorption,
physical models accounting for mobile-immobile water described better reactive solute transport
in a field-scale natural gradient tracer study (Goltz and Roberts, 1986), although other factors
contributing to non-ideal behavior may also be important (Brusseau, 1994).
The development of heterogeneous unsaturated flow, in non-aggregated porous media was
suggested in several studies comparing the transport of non-sorptive tracers at various degrees of
moisture saturation (Biggar and Nielsen, 1962; Bond and Wierenga, 1990; Nielsen and Biggar,
1961). The fraction of immobile water increased from 4 to 40 percent when water content was

2.32
0.0
0.2
0.4
0.6
0.8
1.0
0 20 40 60 80
Moisture Saturation (%)
Fraction Mobile Water
decreased from 71 to 55 percent moisture saturation in a disturbed sand column (Gaudet et al.,
1977). It is important to note one study with contrasting results where a decrease in dispersion
(heterogeneity) was observed when moisture content was reduced from 100 to 97 and 93 percent
(Jardine et al., 1993). This was observed in undisturbed cores and the effect was attributed to the
elimination of macropore flow for the slightly unsaturated conditions. Changes in hydraulic
heterogeneity at lower water contents were not evaluated.
Recent studies of unsaturated sands (Gamerdinger et al., 1998, Gamerdinger and Kaplan, 1999)
confirmed the findings of Gaudet et al. (1977). The development of an increasing fraction of
immobile water (i.e., decreasing N
m
) is illustrated in Figure 2.4 for sandy soils (filled diamond and
square symbols). Data for a fine-textured soil (loamy fine sand, represented by the filled triangle
in Figure 2.4) with a high N
m
is consistent with the conceptual model (above) that water held in
the smaller pores of unsaturated fine-texture soils remains conductive.
Figure 2.4. Development of hydraulic heterogeneity
(decreasing N
m
) in unsaturated, non-
aggregated soils with decreasing moisture
saturation. [Filled diamond and square
symbols represent data for sandy soils from
Gamerdinger and Kaplan (1999) and
Gaudet et al. (1977), respectively. The
filled triangle symbol is data for finer-
textured loamy fine sand from Bond and
Wierenga (1990).]
2.33
The unsaturated flow regime is expected to differ in soils dominated by coarse-textured particles
in contrast to those with fine-textured particles. As shown schematically in Figure 2.3, the flow
regime will consist of film flow along the surfaces of large particles, versus matrix flow through
small pores formed by fine particles. Hydraulic conductivity varies with soil texture and decreases
with decreasing moisture saturation. Hydraulic heterogeneity resulting from a 2-region, mobile-
immobile water, flow domain, increases in unsaturated soils. Limited data suggest a dependence
on soil texture for non-aggregated soils where the heterogeneity is not apparent for saturated
conditions.
In summary, the development of hydraulic heterogeneity in non-aggregated unsaturated soils has
long been identified. However, the implications for the sorptive and transport behavior of
contaminants are unknown. Current transport models account for lower, and spatially variable,
hydraulic conductivity in unsaturated soils. Hydraulic heterogeneity resulting from mobile-
immobile water domains has been considered when modeling transport in saturated systems.
Fewer investigations have considered unsaturated systems.
2.5 Effects of Chemical Heterogeneity on Transport
In context of contaminant adsorption and transport, chemical heterogeneity refers in this report to
the variability in particle surface reactivity. Iron oxide minerals are abundant in the subsurface
environment and mineral coatings consisting of Fe(III)-oxides can be a significant source of
reactivity for contaminant adsorption in a variety of soil systems [e.g., Smith and Jenne (1991) as
summarized by Tompson et al. (1996)]. The effect of chemical heterogeneity arising from
spatially variable Fe(III)-oxide abundance has been considered in modeling the transport of cobalt
(Brusseau and Zachara, 1993) and reactive transport of uranyl-citrate (Tompson et al., 1996) and
Co-EDTA (Szecsody et al., 1998a, 1998b) complexes. Modeling studies of reactive solute
transport in saturated systems have shown that chemical heterogeneities result in non-ideal
transport behavior (Bosma et al., 1993, Sugita et al., 1995).
Different approaches for representing chemical heterogeneity (i.e., spatial distribution of sorption
sites) have been considered when combining processes in reactive flow and transport models for
saturated systems (Szecsody et al., 1998a, 1998b; Tompson et al., 1996). The effect of a
homogeneous versus a heterogeneous (spatially variable) distribution of reactive sites was
compared in simulations of reactivity in batch (non-flowing) and column systems (Szecsody et al.,
1998b). While reactive site heterogeneity had a small effect in batch systems, it was highly
significant in columns. A major conclusion was that particle-scale heterogeneities significantly
influenced the interactions of sorptive (and reactive) solutes during advective flow (Szecsody et
al., 1998b).
2.34
2.5.1 Coupled Hydraulic and Chemical Heterogeneity
The absence of well-controlled laboratory investigations of contaminant transport in physically
and chemically heterogeneous porous media has been noted (Brusseau and Zachara, 1993). The
increased importance of particle scale heterogeneity in flowing versus non-flowing systems has
implications for the advance from saturated to unsaturated solute transport. Variability in
accessibility to reactive sites by mobile solutes is expected to increase with increased tortuosity.
Predictive models that incorporate heterogeneity usually focus on reactive site abundance
(Szecsody et al., 1998a; Thompson et al., 1996), although the length of Fe(III)-oxide inclusions
has been considered (Szecsody et al., 1998a). In an unsaturated soil, the distribution and
abundance of reactive sites is considered to be the same as when the soil is saturated. However,
the observed increase in hydraulic heterogeneity with decreased moisture saturation suggests
greater variability in access to reactive sites, and thus an even greater significance of particle scale
chemical heterogeneity during transport in unsaturated soils. Models which consider spatially
variable reaction capacity and accessibility have been developed for saturated porous media flow
(Reichle et al., 1998).
Unsaturated systems have received less attention, with few investigations considering the
combined effects of hydraulic and chemical heterogeneity. Russo (1989a,b) evaluated spatially
variable hydraulic conductivity from a different perspective, considering the effect of solution
properties on soil properties which included hydraulic conductivity and water content. The spatial
variability of pesticide transport and sorption in unsaturated soil was evaluated in field, laboratory
column, and batch experiments (Elabd et al., 1986; Jury et al., 1986). Although average K
d
values from batch and column experiments were similar, there was no correlation between values
measured in batch (non-flowing) and column systems (Jury et al., 1986). For 18 of the 36
samples, retardation in laboratory columns with undisturbed field cores was greater than predicted
from the batch K
d
value (Elabd et al., 1986). Non-sorptive tracer (e.g., chloride) transport did
not show the same degree of heterogeneity as the pesticides, suggesting sorption variability in
addition to flow heterogeneity (Jury et al., 1986). This research indicates that sorption during
unsaturated transport is not accurately predicted from measurements in batch systems.
An important issue for modeling transport in a chemically heterogeneous system is the correlation
between reactivity and other parameters that are used to characterize the medium. Correlations
between reactivity and permeability (or hydraulic conductivity) are often used, but the appropriate
form of the correlation for specific systems can be debated (Tompson et al., 1996). Data to
support specific correlations between particle size and particle surface reactivity are sparse.
Consequently, approximate negative correlations are often used (Burr et al., 1994; Tompson,
1993; Tompson et al., 1996), or alternative possibilities of negative, positive, or no correlation are
evaluated (Bosma et al., 1993, 1996; Tompson, 1993;).
Experimental evidence for a negative correlation between particle size and the sorption of organic
chemicals has been demonstrated (Barber et al., 1992; Karickhoff et al., 1979; Schwarzenbach
and Westall, 1981). However, the findings of Ball et al. (1990) for the Canadian Forces Base

2.35
J
ix
' & D
dC
i
dx
(2.35)
Borden Aquifer soils do not support this trend. A significant, but weak, correlation between
strontium sorption and “ln K
h
” was observed for the Borden Aquifer soils (Robin et al., 1991).
Preferential sorption of strontium to the fine-textured soil fraction and with micaceous minerals of
the coarsest fraction of Chalk River aquifer soils from Ontario, Canada was observed by Pickens
et al. (1981).
The importance of grain scale properties in determining the sorption and transport of reactive
solutes and the need for systematic study of factors that affect correlation for specific porous
media and reactions is evident. Due to the increasing complexity of unsaturated systems, the
success of simply extending relationships that are developed in saturated systems is questionable.
Fundamental research in unsaturated systems with measurement of relevant parameters relating
media properties to chemical heterogeneity and reactive transport is needed.
In summary, the relationships between soil texture, porosity, moisture saturation, hydraulic
heterogeneity and sorption are complex. Hydraulic heterogeneity in unsaturated soils results from
disconnectivity of pore water which can be caused by thin liquid films around soil particles, dead-
end pores, or relatively isolated regions associated with unsaturated flow. The proportion of
disconnected or immobile water increases with decreasing moisture saturation (Figure 2.4). The
unsaturated flow regime is thought to depend on the soil texture, where film flow is more likely to
dominate in unsaturated coarse- textured soils, and matrix flow through small pores prevailing in
fine-textured soils.
2.6 Diffusion
The transport of matter in the absence of bulk flow is referred to as diffusion. The flux of matter
due to diffusion is proportional to the concentration gradient and is a molecular process. In
general terms, the flux, J
ix
, of component I in the x direction is
where D = proportionality constant or diffusion coefficient with the dimensions of length
2
/time
C
i
= concentration of constituent i.
The J
ix
has the dimensions of moles/length
2
/time, and the negative sign indicates that flow of
constituent I in the x direction is also in the direction of lower I concentration. In an infinitely
dilute aqueous solution, the movement is quantified by the diffusion coefficient, D. For most
simple aqueous species, D is about 10
-9
m
2
/s or 10
-5
cm
2
/s.
Atkinson (1983), Atkinson et al. (1986), and Atkinson and Nickerson (1988) present a useful
conceptual model for describing the transport of contaminants through a porous media such as
soil. The authors consider that the transport is a combination of both physical processes, such as
diffusion, and chemical processes, such as precipitation/solubility and adsorption/desorption. In

2.36
D
p
'
D *
J
2
(2.36)
D
i
' D
p
n
e
'
D n
e
*
J
2
(2.37)
D
a
'
D
i
"'
'
D
p
n
e
"'
'
D n
e
*
J
2
"'
(2.38)
"' ' n
e
% D
b
K
d
(2.39)
"'
n
e
' R
f
' 1 %
D
b
K
d
n
e
(2.40)
the constrained geometry of a porous media, such as soil, the D is reduced compared to the D in
free aqueous solution. The D for a species within a porous media is defined as D
p
and is equal to
where * = constrictivity of the porous media
J = tortuosity of the porous media.
For experimentalists, it is convenient to measure the average flux of a contaminant per unit area of
the porous media in relation to the concentration gradient of the contaminant in the aqueous
phase. The concentration gradient in the aqueous phase is influenced by the volume fraction of
the void space in the porous media (the porosity, n
e
). This leads to another equation that defines
the "intrinsic" diffusion coefficient, D
i
:
A key assumption herein is that all the porosity in the porous media is interconnected and thus can
contribute to diffusion of the contaminant. All three parameters, porosity (n
e
), constrictivity (*),
and tortuosity (J), characterize the physical contribution to diffusion through the porous media.
The chemical contributions to diffusion can potentially be quite varied, such as ion exchange,
specific adsorption, precipitation, and lattice substitution. If a very simple chemical process is
assumed, reversible surface adsorption having fast kinetics and a linear isotherm (i.e., K
d
), then
diffusion of a reactive contaminant can be characterized by an apparent diffusion coefficient, D
a
:
where "' = capacity factor or ratio of the moles per unit volume of water-saturated solid,
C
s
, to the moles per unit volume of liquid, C
l
.
The capacity factor is related to the K
d
by the equation
where D
b
= dry bulk density of the porous media.
Also note that "'/n
e
is the familiar retardation factor used in transport modeling:

1
A colloid is any fine-grained material, sometimes limited to the particle-size range of
<0.00024 mm (i.e., smaller than clay size), that can be easily suspended (Bates and Jackson,
1979). In its original sense, the definition of a colloid included any fine-grained material that does
not occur in crystalline form. The geochemistry of colloid systems is discussed in detail in sources
such as Yariv and Cross (1979) and the references therein.
2.37
MC
Mt
'
D
R
f
M
2
C
M x
2
(2.41)
It should be noted that these simple relationships are strictly only valid for reversible, linear
adsorption reactions with fast kinetics. This point is often overlooked and should not be.
Nevertheless, such simplifying assumptions allow for some interesting analysis of common
laboratory data and experiments.
Equation 2.40 has been incorporated into the transport equation:
where R
f
= retardation factor, as defined in Equations 2.23 or 2.24
t = time.
Note that Equation 2.41, which is strictly for transport by diffusion, is similar to Equation 2.25,
which describes contaminant migration due to advective flow.
Therefore, the apparent diffusion coefficients, D
a
, for reactive constituents account for the
chemical retardation as well as the physical hindrance to contaminant mobility caused by the small
pore sizes and tortuosity of the soil.
2.7 Subsurface Mobile Colloids
2.7.1 Concept of 3-Phase Solute Transport
Contaminant transport models generally treat the subsurface environment as a 2-phase system in
which contaminants are distributed between a mobile aqueous phase and an immobile solid phase
(e.g., soil). Contaminants with a high affinity for sorbing to rock or vadose zone soils are
assumed to be retarded relative to the rate of groundwater flow. However, an increasing body of
evidence indicates that under some subsurface conditions, components of the solid phase may
exist as colloids
1
that may be transported with the flowing water. Association of contaminants
with this additional mobile phase may enhance not only the amount of contaminant that is
transported, but also the rate of contaminant transport. Most current approaches to predicting
contaminant transport ignore this mechanism not because it is obscure or because the
mathematical algorithms have not been developed (Corapcioglu and Kim, 1995; Mills et al.,
1991), but because little information is available on the occurrence, the mineralogical properties,
the physicochemical properties, or the conditions conducive to the generation of mobile colloids.
2.38
There are 2 primary problems associated with studying colloid-facilitated transport of
contaminants under natural conditions. First, it is difficult to collect colloids from the subsurface
in a manner which minimizes or eliminates sampling artifacts. Sampling artifacts can arise when
groundwater is pumped too rapidly, yielding particles that would otherwise remain immobile in
the aquifer (Backhus et al.,1993; McCarthy and Degueldre, 1993; Powell and Puls, 1993).
Colloids may also be generated during sampling by exposing groundwater containing readily
oxidizable metals, such as Fe(II), to atmospheric conditions and causing the precipitation of fine
grain-sized hydrous oxide colloids (Backhus et al., 1993; McCarthy and Degueldre, 1993; Ryan
and Gschwend, 1990). Secondly, it is difficult to unambiguously delineate between the
contaminants in the mobile-aqueous and mobile-solid phases (Buffle et al., 1992; Degueldre et al.,
1989; McCarthy and Degueldre, 1993; Puls, 1990). Using ultrafiltration techniques to accomplish
this goal is not entirely satisfactory because it provides only indirect evidence, is subject to a
number of artifacts, and usually requires high analytical precision at very low contaminant
concentrations (Buffle et al., 1992; Danielsson, 1982; Degueldre et al., 1989; McCarthy and
Degueldre, 1993).
2.7.2 Sources of Groundwater Mobile Colloids
Subsurface mobile colloids originate from (1) the dispersion of surface or subsurface soils,
(2) decementation of secondary mineral phases, and (3) homogeneous precipitation of ground-
water constituents (McCarthy and Degueldre, 1993). First, colloidal particles can be dispersed
and become mobile in aquifers as a result of changes in the groundwater chemistry, such as a
decrease in ionic strength or changes in ionic composition from a calcium- to a sodium-dominated
chemistry. The effect of sodium and ionic strength on colloid suspension stability is interactive
such that the dispersive quality of sodium is enhanced at low salt levels (Kaplan et al., 1996).
Geochemical or microbiological changes that result in dissolution of cementing phases, such as
iron oxides and calcium carbonate can result in release of colloids. For example, Gschwend et al.
(1990) observed 10 to 100 mg/l of silica colloids in groundwater receiving recharge from
evaporation ponds and a fly ash basin. The infiltrate was enriched in carbon dioxide that dissolved
the soil-cementing carbonate mineral, thus releasing the silica colloids. The third source of
groundwater mobile colloids is homogeneous precipitation. Changes in groundwater geochemical
conditions such as pH, major element composition, redox potential, or partial pressures of CO
2
can induce supersaturation and coprecipitation of colloidal particles. The precipitates can include
major elements such as oxides of iron and manganese, calcium carbonates, and iron sulfides, as
well as minor elements such as carbonates and sulfides of metals and radionuclides. Mobile
colloid precipitates may form when soluble contaminants are introduced into a system resulting in
their exceeding the solubility product. For example, Gschwend and Reynolds (1987) observed
precipitation of ferrous phosphate colloids (1 to 10 mg/l of 100 nm-sized particles) down gradient
of a sewage infiltration site. Solubility calculations suggested that the dissolved phosphate ions
from the sewage and reduced iron in the aquifer exceeded the solubility product of ferrous
phosphate, resulting in the formation of insoluble colloids. Other studies have documented the
formation of iron oxide colloids in groundwater as a result of changes in pH and oxygenation that
2.39
caused the solubility limit of Fe(III) oxides to be exceeded (Liang et al., 1993). Many strongly
hydrolyzing radionuclides also form submicron-sized particles. Increases in solution pH have
been shown to induce the formation of plutonium-, uranium-, and americium-oxide colloids in
carbonate systems (Ho and Miller, 1986; Kim, 1986).
2.7.3 Case Studies of Mobile-Colloid Enhanced Transport of Metals and Radionuclides
Although the concept of colloid-facilitated transport is often invoked to account for anomalies
between predicted and observed transport of contaminants, little field or experimental verification
of this potentially important phenomenon is available (McCarthy and Degueldre, 1993). There
have been a few field studies describing colloid-facilitated transport and these studies have
provided only circumstantial evidence that this is in fact the actual mechanism responsible for the
enhanced transport of contaminants (Buddemeier and Hunt, 1988; Degueldre et al., 1989; Kaplan
et al., 1994a; Kaplan et al., 1995a; Penrose et al., 1990).
Although laboratory studies at Los Alamos National Laboratory (LANL) predicted that the
movement of actinides in subsurface environments would be limited to less than a few meters,
both americium and plutonium were detectable in monitoring wells as far as 3,390 m down
gradient from the point source (Penrose et al., 1990). Almost all of the americium and plutonium
in the groundwater at the 3,390 m well were associated with colloids 0.025 to 0.45 µm in
diameter. Similarly, based on laboratory measurements using site-specific soils and a 2-phase
solute transport code, americium, curium, plutonium, and uranium were expected to travel less
than 10 m in the F-Area of the Savannah River Site; the contaminants were found associated with
groundwater colloids 1,200 m away from the point source (Kaplan et al., 1994a). To a lesser
extent, chromium, copper, nickel, and lead were also detected directly on suspended groundwater
particles collected from the F-Area study site (Kaplan, 1994b, 1995a). The reader is cautioned
that, because most transport predictions do not account for preferred flow paths, the
interpretation that colloid-facilitated migration is the best explanation for such enhanced migration
is still being debated.
2.8 Anion Exclusion
Dissolved chloride, bromide, and nitrate are usually reported to travel through natural systems or
soil columns at the same rate as, or faster than, water ( James and Rubin, 1986; McMahon and
Thomas, 1974). Anion exclusion, the mechanism by which anions move faster than water, occurs
when the diffuse double layer, an extension of a particle's negative surface charge into the
surrounding solution, repulses anions (Sposito, 1984). By excluding anions from the diffuse
double layer, where water is relatively immobile, the system restricts anions to the faster moving
pore water, resulting in an average rate of anion transport that is greater than the average pore
water velocity defined by Darcy’s Law (James and Rubin, 1986; McMahon and Thomas, 1974).
Anion exclusion is more pronounced with higher cation exchange capacity (i.e., negative charge)
of the soil or rock. For example, smectites (CEC.3 meg/g) exhibit anion exclusion to a greater
degree than do the kaolinite (CEC.0.2 meg/g) minerals (McMahon and Thomas, 1974).
2.40
The implication of anion exclusion for anionic contaminants (e.g., nitrates, chloride, chromate,
pertechnetate) and anionic complexes [e.g., UO
2
(CO
3
)
2
2-
] is that they may be able to travel through
the subsurface at a rate greater than water. There is some indirect evidence that anion exclusion
may exist for pertechnetate. In an unsaturated column study, pertechnetate breakthrough (C/C
0
,
0.5) occurred at 0.95 pore volumes, whereas for tritium, a conservative tracer, breakthrough
occurred at 1.02 pore volumes; that is, pertechnetate may have traveled 5 percent faster than
tritium (Gee and Campbell, 1980). Chloride breakthrough in these columns occurred at 0.80 pore
volumes, or 22 percent faster than tritium, providing evidence that the transport of chloride may
be affected by anion exclusion.
2.9 Summary
The objective of this chapter is to present a primer on the key geochemical processes affecting
contaminant transport in subsurface environments. References to important review articles and
books are included for each of the major subject areas: aqueous geochemical processes, sorption,
diffusion, subsurface mobile colloids, and anion exclusion. These processes are summarized in
Table 2.5.
Particular attention is directed at describing the geochemical processes affecting chemical
retardation. A brief discussion of mechanistic and semi-empirical adsorption models.
Incorporating mechanistic and semi-mechanistic adsorption concepts into transport models is
desirable because the models become more robust and scientifically defensible. However,
mechanistic models are rarely, if ever, applied to field-scale problems. The reasons for this are the
following: (1) mechanistic models require a more intense data collection effort than will likely be
available to the majority of transport modelers, licensee requestors, or responsible parties; (2)
installing these mechanistic adsorption models into existing transport codes is quite complex; and
(3) mechanistic adsorption models require full characterization of the mineral surfaces,
information that is impossible to obtain in natural heterogeneous soils. Importantly, these models
provide a paradigm for using simpler models, such as the conditional K
d
model.
Conditional K
d
values can be derived from laboratory or field experiments. Unlike the
thermodynamic K
d
term, they are less rigorously defined in that the conditional K
d
values are not
necessarily limited to a single aqueous species and single solid phase. This broader definition
lends itself more readily to natural systems, while at the same time resulting in several technical
issues and complexities. The understanding of the important geochemical factors affecting the
transport of the contaminants of interest is critical for site-specific calculations.

2.41
Table 2.5. Summary of chemical processes affecting attenuation and mobility of contaminants.
Process Mechanism
Enhancement of
Attenuation or
Mobility of
Contaminant?
Key Facts
Aqueous
Complexation
Reaction where an
aqueous molecular unit
(ion) acts as a central
group to attract and
form a close
association with other
atoms or molecules
May enhance
attenuation or mobility,
depending on
contaminant and
geochemical conditions
C Function of pH and redox
C Complexation may lower the
potential for adsorption and/or
increase solubility, both of which
can enhance potential for mobility
C Complexes may more readily bond
to soils and thus retard migration
C Organic ligands from humic
materials can be present in
significant concentrations and
dominate contaminant
complexation in some systems
Redox Reactions Reaction where
electrons are
transferred completely
from one species to
another
May enhance
attenuation or mobility,
depending on
contaminant and
geochemical conditions
C Change in redox status changes
aqueous speciation which may
increase or decrease adsorption
and solubility
C If redox status is sufficiently low to
induce precipitation of sulfide
minerals, reprecipitation of some
contaminants may be expected
C More difficult to predict mobility
of redox-sensitive species because
many redox reactions are kinet-
ically slow in natural groundwater,
and several elements may never
reach equilibrium between their
various valence states
2.42
Table 2.5. Continued.
Process Mechanism
Enhancement of
Attenuation or
Mobility of
Contaminant?
Key Facts
Adsorption and
Ion Exchange
Special case of a
complexation reaction
where there is a net
accumulation of a
contaminant at the
interface between a
solid phase and an
aqueous-solution
phase; does not include
the development of a
3-dimensional
molecular structure
Enhances Attenuation C Occurs primarily in response to
electrostatic attraction
C Very dependent on pH and
mineralogy
C Anion adsorption is greatest at low
pH and decreases with increasing
pH
C Cation adsorption is greatest at
high pH and decreases with
deceasing pH.
C Some contaminants may be
present as cations or anions
depending pH
C Totally-to-partially reversible;
decline in contaminant
concentration in groundwater may
result in desorption and release of
adsorbed contaminant to
groundwater
C Likely key process controlling
contaminant mobility in areas
where chemical equilibrium exists
Precipitation Special case of a
complexation reaction
in which the complex
formed by 2 or more
aqueous species is a
solid with
3-dimensional
molecular structure
Enhances Attenuation C Very dependent on pH and redox
C Totally-to-partially reversible;
decline in contaminant
concentration in groundwater may
result in dissolution of precipitated
contaminant to groundwater
C Likely process where chemical
nonequilibium exists, an area
where high contaminant
concentrations exist, or where
steep pH and/or redox gradients
exist
2.43
Table 2.5. Continued.
Process Mechanism
Enhancement of
Attenuation or
Mobility of
Contaminant?
Key Facts
Diffusion Molecular process of
transport of matter in
the absence of bulk
flow
Enhances Mobility C Flux of matter due to diffusion is
proportional to concentration
gradient
Subsurface
Colloids
Contaminants
associated with
suspended fine-grained
material (smaller than
clay size) that may be
transported with
flowing groundwater
Enhances Mobility C Little information on occurrence,
mineralogical and
physicochemical properties, or
conditions conducive to the
generation of mobile colloids
C May originate from the dispersion
of soils, decementation of
secondary mineral phases, and/or
precipitation of groundwater con-
stituents
C Difficult to collect colloids from
subsurface in a manner that
minimizes or eliminates sampling
artifacts
C Difficult to unambiguously
delineate between the
contaminants in the mobile-
aqueous and mobile-solid phases
Anion Exclusion Occurs when the
diffuse double layer, an
extension of a particle's
negative surface charge
into the surrounding
solution, repulses
anions
Enhances Mobility C By excluding anions from the
diffuse double layer, where water
is relatively immobile, anions
restricted to the faster moving pore
water, resulting in an average rate
of anion transport greater than the
average pore water velocity
defined by Darcy’s Law
C more pronounced with higher
cation exchange capacity (i.e.,
negative charge) of the soil or rock

1
A list of acronyms, abbreviations, symbols, and notation is given in Appendix A. A list of
definitions is given in Appendix B
3.1
3.0 Methods, Issues, and Criteria for Measuring K
d
Values
3.1 Introduction
The partition (or distribution) coefficient, K
d
,
1
is a measure of sorption of contaminants to soils
and is defined as the ratio of the quantity of the adsorbate adsorbed per unit mass of solid to the
amount of the adsorbate remaining in solution at equilibrium. It is the simplest, yet least robust
model available. There are 5 general methods used to measure K
d
values: laboratory batch
method, in-situ batch method, laboratory flow-through (or column) method, field modeling
method, and K
oc
method. Each method has advantages and disadvantages, and perhaps more
importantly, each method has its own set of assumptions for calculating K
d
values from
experimental data. Consequently, it is not only common, but expected that K
d
values measured by
different methods will produce different values.
A number of issues exist concerning the measurement of K
d
values and the selection of K
d
values
from the literature. These issues include: using simple versus complex natural geologic materials
as adsorbents, field variability, the “gravel issue,” the “colloid issue,” and the particle
concentration effect. Soils are a complex mixture containing solid, gaseous, and liquid phases.
Each phase contains several different constituents. The use of simplified systems containing single
mineral phases and aqueous phases with 1 or 2 dissolved species have provided valuable
paradigms for understanding sorption processes in more complex, natural systems. However, the
K
d
values generated from these simple systems are generally of little value for importing directly
into transport models. Values for transport models should be generated from materials from or
similar to the study site. The “gravel issue” is the problem that transport modelers face when
converting laboratory-derived K
d
values based on experiments using the less than 2-mm fraction
into values that can be used in systems containing particles greater than 2 mm in size. No
standard methods exist to address this issue. The “colloid issue” was discussed previously in
Section 2.7. Some investigators have observed that K
d
values determined in the laboratory often
decrease as the ratio of solid to solution used in the measurements increases. This particle
concentration effect is puzzling, because a K
d
value should not depend from a theoretical
perspective on the solid-to-solution ratio. Investigators have offered several explanations
involving physical/chemical processes and/or experimental artifacts for the observed dependency.
Spatial variability provides additional complexity to understanding and modeling contaminant
retention to subsurface soils. The extent to which contaminants partition to soils often changes as
field mineralogy and chemistry changes. Thus, a single K
d
values is often not sufficient for an
entire study site and should change as important environmental conditions change. It is therefore
important to be able to identify and measure the effect of ancillary environmental parameters that
influence contaminant sorption. Three approaches used to vary K
d
values in transport codes are
the K
d
look-up table approach, the parametric K
d
approach, and the mechanistic K
d
approach.

3.2
A % C
i
' A
i
,
(3.1)
K
d
'
A
i
C
i
(3.2)
The extent to which these approaches are presently used and the ease of incorporating them into
flow models varies greatly.
The objective of this chapter is to provide an overview of the different methods of measuring and
determining K
d
values used in site-specific contaminant transport and risk assessment calculations.
Issues regarding the selection of K
d
values from the literature for use in screening calculations are
discussed.
3.2 Methods for Determining K
d
Values
There are 5 methods of determining K
d
values: (1) laboratory batch method, (2) in-situ batch
method, (3) laboratory flow-through (or column) method, (4) field modeling method, and (5) K
oc
method (EPA, 1991; Ivanovich et al., 1992; Jackson and Inch, 1989; Johnson et al., 1995;
Karickhoff et al., 1979; Landstrom et al., 1982; Lyman et al., 1982; Roy et al.; 1991; Serkiz et
al., 1994; Sposito, 1984; van Genuchten and Wierenga; 1986). Each method provides an
estimate of the propensity of a contaminant to sorb to the solid phase. However, the techniques
used and the assumptions underlying each method are quite different. Consequently, K
d
values
for a given system that were measured by different methods commonly have values ranging over
an order of magnitude (Gee and Campbell, 1980; Relyea, 1982). This subsection will describe the
different methods and compare their implicit and explicit assumptions.
The K
d
model originates from thermodynamic chemistry (see detailed discussion in Chapter 2)
(Alberty, 1987). It is a measure of sorption and is defined as the ratio of the quantity of the
adsorbate adsorbed per gram of solid to the amount of the adsorbate remaining in solution at
equilibrium. For the reaction
the mass action expression is the partition coefficient (K
d
, ml/g):
where A = concentration of free or unoccupied surface adsorption site on a solid phase
(mol/ml),
C
i
= total dissolved adsorbate concentration remaining in solution at equilibrium
(mol/ml or µg/ml), and
A
i
= concentration of adsorbate on the solid at equilibrium (mol/g or µg/g).
Equation 3.2 is valid only when A is in great excess with respect to C
i
and the activity of A
i
is
equal to unity. For saturated conditions and non-polar organic constituents, sorption from the
aqueous phase to the porous media of the subsurface can be treated as an equilibrium-partitioning

3.3
A
i
' q
i
'
V
w
(C
0
& C
i
)
M
sed
(3.3)
K
d
'
V
w
(C
0
& C
i
)
M
sed
C
i
(3.4)
process when solute concentrations are low (e.g., either #10
-5
molar, or less than half the
solubility, whichever is lower) (EPA, 1989). Partitioning often can be described using the above
linear isotherm.
Also inherent in the thermodynamic definition of the K
d
term are the assumptions that the reaction
is independent of the contaminant concentration in the aqueous phase and that the system is
reversible, i.e., that the desorption rate is equal to the adsorption rate. The thermodynamic K
d
term describes a precisely defined system, including fixed pH and temperature, with one type of
adsorption site, A, and one type of dissolved aqueous species, C
i
. Although the thermodynamic
K
d
term is overly restrictive for use in natural heterogeneous systems, it provides an important
paradigm to base empiricised K
d
terms. The assumptions that need to be made to empiricise this
construct vary between analytical methods.
3.2.1 Laboratory Batch Method
Batch studies represent the most common laboratory method for determining K
d
values (ASTM,
1987; EPA, 1991; Roy et al., 1991). Figure 3.1 illustrates an EPA (1991) procedure for
measuring a batch K
d
value. A well characterized soil of known mass (M
sed
) is added to a beaker.
A known volume (V
w
) and concentration (C
0
) of an aqueous contaminant solution is added to the
soil in the beaker. The beaker is sealed and mixed until sorption is estimated to be complete,
typically 1 to 7 days. When possible, the person conducting the study should ascertain the actual
time required to reach sorption equilibrium. The solutions are centrifuged or filtered, and the
remaining concentration of the contaminant (C
i
) in the supernatant is measured. The
concentration of adsorbate sorbed on the solid phase (A
i
, sometimes noted as q
i
) is then calculated
by Equation 3.3:
Equation 3.3 is used to calculate the numerator of the K
d
term (Equation 3.2) and the
denominator, C
i
, of the K
d
term is measured directly in the laboratory. Thus,
For organic compounds that can degrade into other compounds, it should be noted that the
difference in solution concentrations in Equation 3.3 represents both adsorption and degradation.
Therefore, the calculated K
d
for organic compounds of this type can overestimate the amount of
true adsorption. If container blanks are not included in the batch test matrix, adsorption of a
contaminant to the container is included in the calculated K
d
. Care must be taken when
interpreting batch K
d
test results.

3.4
Figure 3.1. Procedure for measuring a batch K
d
value (EPA, 1991).
It is important to note that the interpretation of results from batch K
d
sorption tests generally
allow no distinction to be made on how the sorbate (i.e., contaminant) is associated with the
sorbent (i.e., soil). The sorbate may be truly adsorbed by ion exchange, chemisorption, bound to
complexes that are themselves sorbed on the solid, and /or precipitated. If the K
d
values are going
to be used in transport calculations that already account for precipitation processes, it is
imperative that the K
d
values only include the decrease in dissolved concentrations of the sorbate
due to adsorption. That is, the user must be certain that the experiments were performed
correctly to prevent significant removal of the sorbate by precipitation reactions. Otherwise, the
estimated retardation can be significantly overestimated.
There are several variations of this general procedure, each variation addressing the specific needs
of the system. It is necessary to have some latitude in the method because of limits due to
analytical chemistry considerations. For instance, for contaminants in which very low sorption is
expected, a larger ratio of solid to liquid may increase the small difference in the term (C
0
- C
i
).
Conversely, for contaminants in which high sorption is expected, a lower ratio of solid to liquid
may be desirable. For gamma-ray emitting contaminants, it is possible to directly count the
3.5
activity on the equilibrated solid and in the solution, such that the K
d
can be directly determined as
opposed to relying on the difference in activity (i.e., concentration) in the solution phase only.
One of the most common variations of the EPA method is to conduct a series of batch tests that
are identical except for varying of the concentration of the dissolved contaminant, C
i
. The K
d
for
the resulting isotherm is typically calculated from the slope of a C
i
versus A
i
plot. As discussed in
Section 2.3.3, adsorption isotherm experiments are often conducted to evaluate the effect of
contaminant concentration on adsorption, while other parameters are held constant. For soils, it
is common knowledge that contaminant adsorption can deviate from the linear relationship
required by the K
d
construct. This approach obviously requires more work, but can provide a
more accurate estimate.
Other variations of the batch K
d
procedure deal with the ratio of solids to liquid, liquid
composition, and contaminant concentration. A detailed detailed description of a batch K
d
procedure is included in Appendix C.
Contaminant transport modelers are often interested in the K
d
value of a contaminant in a specific
groundwater plume (e.g., an acidic plume) in contact with a specific soil. In such a case, an
experimenter would spike the contaminant into a representative groundwater, as opposed to pure
water. Additionally, the experimenter would attempt to equilibrate the soils with the background
aqueous solution (e.g., the acidified groundwater) before bringing the soil in contact with the
contaminant of interest. The reason for this latter step is to isolate the adsorption/desorption
reaction of interest between the contaminant and soil. By pre-equilibrating the soil first with the
acidic plume water (without the contaminants present), all the extraneous chemical reactions
should be near equilibrium. Then, when the contaminant is added, its reaction is isolated.
The batch method is popular because the equipment, cost, and time requirements are low and the
methodology is quite simple. However, the seemingly elementary operations mask numerous
subtleties resulting in variability of data (EPA, 1991; Roy et al., 1991; Serne and Relyea, 1981).
One of the most comprehensive exercises to evaluate interlaboratory precision and identify
important procedural details was conducted by 9 laboratories (Serne and Relyea, 1981). General
guidelines on groundwater compositions, radionuclides, and procedural details were given to
participants in this exercise. The measured K
d
values were surprisingly varied for 2 of
3 contaminants investigated. As much as 3 orders of magnitude difference were determined in
cesium (1.3 ± 0.4 to 880 ± 160 ml/g) and plutonium (70 ± 36 to 63,000 ± 19,000 ml/g).
Conversely, the strontium K
d
values measured in the 9 laboratories were within an order of
magnitude of each other, 1.4 ± 0.2 to 14.9 ± 4.6 ml/g. Serne and Relyea (1981) concluded that
the cause of the variability of the plutonium and cesium K
d
values was due to: (1) method of
tracer addition to solution, (2) solution-to-solid ratio, (3) initial tracer concentration in influent
solution, (4) particle size distribution, (5) solid-solution separation method, (6) sample containers,
and (7) temperature. The authors discussed in detail each of these parameters that are generally
not controlled in batch K
d
methods.
3.6
Essentially all of the assumptions associated with the thermodynamic K
d
value (Equation 3.2) are
violated in the common batch K
d
value. The natural soils used in these studies are not completely
defined or quantified with respect to their mineralogy and organic phases. The background
aqueous phases that are spiked with the adsorbate are typically not pure water and are rarely
completely characterized, especially in the case when natural groundwater are used as the
background aqueous phase. The background aqueous phases often contain the dominant
electrolytes of the study site or actual uncontaminated groundwater from the study site, consisting
of several dissolved and perhaps colloidal species. Furthermore, the sorption/desorption process
of adsorbates from soils is typically not reversible, i.e., hysteresis is observed, such that desorption
occurs at a slower rate than sorption (Sposito, 1994). However, the batch K
d
term can be of
much greater value to the contaminant transport modeler than the thermodynamic value if the soil
and the aqueous phase closely represent the natural system being modeled. Importantly, such a
complex system, though not completely characterized, provides the best available estimate of the
extent to which a sorbate partitions to a given soil in the presence of the electrolytes present in the
experiment. This issue of measuring K
d
values in complex- versus simple-systems is further
discussed in Section 3.3.1.
One significant limitation inherent in the batch method is that commonly used analytical
instruments can not differentiate between species of a given contaminant. For example, the
atomic absorption (AA) spectrophotometer can measure total cadmium in the aqueous phase but
can not identify each of its species [e.g., Cd
2+
, CdSO
4
"
(aq), CdCl
-
, etc.]. Multiple species typically
exist in groundwater and the effect of their individual K
d
values have a profound effect on the
overall K
d
value. For example, consider a system that consists of a contaminant or radionuclide
with 2 equal concentration species that are kinetically slow at converting between each
composition state; one with a K
d
of 0 ml/g and the second with a K
d
of 1,000 ml/g. The
laboratory batch method would yield an intermediate K
d
of about 30 ml/g in an experiment with a
solution-to-solid ratio of 30. A demonstration calculation illustrating this issue is given in
Figure 3.2. Using the K
d
value 30 ml/g in subsequent mass transport calculations would not be
conservative because 50 percent of the radionuclide would move at the speed of the carrier
solution. For this reason, when there is any suspicion that multiple species with significantly
differing K
d
values may be present, a second sorption methodology, such as the flow-through
method (Section 3.2.3), should be run to search for early breakthrough.

3.7
K
d
'
V
w
(C
0
& C
i
)
M
sed
C
i
C
Xi
'
C
0,X
V
w
K
d,X
M
sed
%V
w
C
Bi
'
500 x 30
(1,000 x 1) % 30
'
15,000
1,030
' 14.56
C
Ai
% C
Bi
' 500 % 14.56 ' 514.56
K
d
'
1,000 & 514.56
514.56
30
1
' 28.30 . 28
Assumptions:
· Total concentration, C
0
, of contaminant I in the original solution is 1,000 mg/ml.
· Batch test is performed with l g of clay soil contacting 30 ml of the original solution.
· The total concentration C
0
is equally divided between two species, A and B, of
contaminant I.
· The true K
d
values for species A and B are 0 and 1,000 ml/g, respectively.
· Kinetic barriers exist that affect their interconversion between these two composition
states over the time period of the test.
Equations and Calculation:
Rearranging the equation
to solve for the concentration of species X of contaminant I (C
Xi
) (i.e., C
Ai
and C
Bi
, where C
A
and C
B
at end of test), one gets
For C
Ai
, we know that there is no adsorption. Therefore, C
Ai
= C
0,A
= 0.5·C
0
= 500 mg/ml.
For C
Bi
, we calculate from the above equation for C
Xi
:
C
i
for total solution is
Therefore, if one does not realize that multiple contaminant species are present which do not
rapidly interconvert, the overall K
d
for the total contaminant would be
Figure 3.2. Demonstration calculation showing affect on overall K
d
by multiple species that
have different individual K
d
values and are kinetically slow at interconverting
between each composition state.
3.8
3.2.2 In-situ Batch Method
A method developed out of the desire to produce an in-situ K
d
value has been used to a limited
extent (Jackson and Inch, 1989; Johnson et al., 1995; Landstrom et al., 1982; McKinley and
Alexander, 1993; Read et al., 1991). The procedure used in this method is somewhat similar to
that of the laboratory batch K
d
method described in Section 3.2.1. A core sample containing a
paired solid and aqueous phase is removed directly from an aquifer. The aqueous phase is
separated from the solid phase by centrifugation or filtration and then analyzed for the solute
concentration, C
i
. The solid is than analyzed for the concentration of the contaminant associated
with the solid phase, A
i
.
Clearly, the advantage of this approach compared to the laboratory K
d
method is that the precise
solution chemistry and solid phase mineralogy is used for the modeling. Furthermore, the pore
water removed from the core material may have had sufficient time to equilibrate and therefore
true equilibrium may been attained. The disadvantages are somewhat less apparent but none the
less appreciable. The concentration of most metal contaminants on the soil surfaces is typically
quite low, in the mg/kg range. It should be noted moreover that the minimum detection limit for
radionuclides on solid surfaces is even lower. The most common instruments available to
measure metal concentrations on surfaces, energy dispersive x-ray analysis (EDX), or x-ray
fluorescence, typically has detection limits in the order of 10,000 and 100 mg/kg, respectively.
Another method of measuring A
i
is to dissolve the solid phase with acid and then measure the
resulting solution by inductively coupled plasma spectroscopy (ICP), inductively coupled
plasma/mass spectroscopy (ICP/MS), and/or atomic adsorption spectroscopy (AA) techniques.
This latter technique may provide a lower (i.e., better) detection limit. In addition to the
detection limit problem, it is not possible by any of these methods to distinguish between sorption
and precipitation - processes which are treated quite differently in transport models. Furthermore,
some trace metals are present in crystalline lattice sites of minerals present in soils. These
molecules are not readily controlled by adsorption/desorption and should not be included in the q
i
term. An in-depth discussion of the limitations of the in-situ batch method is presented by
McKinley and Alexander (1993). For anthropogenic radionuclides present at trace levels, it is
possible to assume that precipitation and lattice site contributions are nil and that the total
mass/activity measured on the solid does represent adsorption/desorption-controlled molecules.
In this scenario, a field in-situ K
d
may be accurate.
One rather successful application of this technique was recently reported by Johnson et al. (1995).
They compared laboratory and field batch K
d
values of uranium along a transect through a pH
gradient of pH 3.0 to 5.6. The field results yielded K
d
values that ranged from 0.4 to greater than
15,000 ml/g for approximately 36 samples. The K
d
values generated by the laboratory batch
technique were generally lower, ranging from 0.08 to greater than 10,000 ml/g. The K
d
values
determined by both methods varied as a function of soil pH at the study site. When both sets of
values were incorporated into a transport code, the results were not significantly different, i.e.,
both methods were essentially equally good at predicting contaminant retardation in the study site.

3.9
3.2.3 Laboratory Flow-Through Method
The laboratory flow-through (or column) method of determining K
d
values is the second most
commonly used method (EPA, 1991; Relyea, 1982; Van Genuchten and Wierenga, 1986).
A solution containing known amounts of a contaminant is introduced into a column of packed soil
of known bulk density (i.e., mass of soil per unit volume of column, g/ml) and porosity (i.e.,
volume of pore space per unit volume of column, ml/ml) (Figure 3.3). The effluent concentration
is monitored as a function of time. A known amount of a nonadsorbing tracer may also be
introduced into the column and its time-varying concentration provides information about the
pore-water velocity. The resulting data is plotted as a break-through curve (Figure 3.3). The
velocity of each constituent (i.e., tracer and contaminant) is calculated as the length of the column
divided by the constituent’s mean residence time.
Figure 3.3. Procedure for measuring a column K
d
value.

3.10
t
pulse
'
t
max
t
min
t C
i
dt
t
max
t
min
C
i
dt
(3.5)
t
step
'
C
max
C
min
t dC
C
max
C
min
C
i
dt
(3.6)
The mean residence time for a pulse input is calculated as follows (Relyea, 1982):
where t
pulse
= mean residence time for a pulse input (hr), t
max
is the end of the break-through
curve (hr),
t
min
= beginning of the break-through curve (hr),
C
i
= constituent concentration [(g or curies)/ml], and
t = time (hr).
The relative concentrations of a constituent at the input source and in the effluent based on a
pulse input are shown schematically in the top left and right of Figure 3.4. The mean residence
time for a step (continual steady-state) input is calculated as follows:
where t
step
= mean residence time for a step input/release (hr),
C
max
= maximum concentration measured in the effluent [(g or curies)/ml], and
C
min
= minimum concentration measured at the beginning of breakthrough [(g or
curies)/ml].
When the effluent curve is ideal, t
step
equals the time when the breakthrough curve reaches 0.5 or
50 percent breakthrough (i.e., C
i
/C
o
=0.5). The relative concentrations of a constituent at the
input source and in the effluent based on a step input are shown schematically in the bottom left
and right of Figure 3.4.
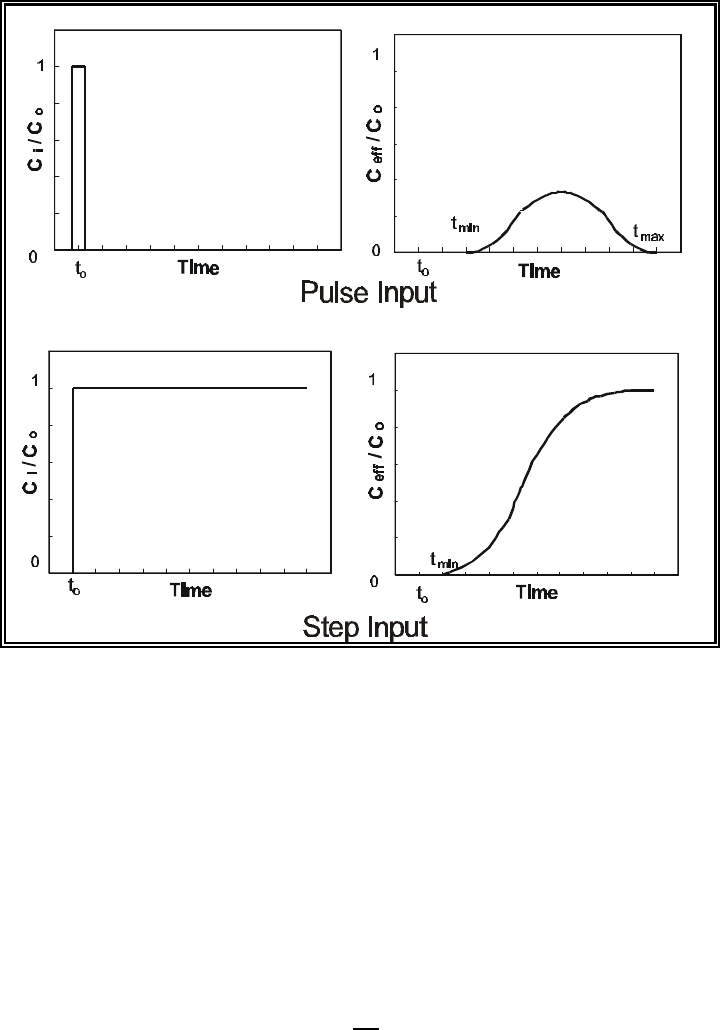
3.11
R
f
'
v
p
v
c
(3.7)
Figure 3.4. Schematic diagram showing the relative concentrations of a
constituent at the input source (figures on left) and in the
effluent (figures on right) as a function of time for a pulse
versus step input. [C
o
, C
i
, and C
eff
refer, respectively, to the
concentration of the constituent at t
o
and the concentrations
of the constituents in the input and effluent.]
The retardation factor (R
f
) is the ratio of the pore-water velocity (v
p
, cm/hr) to the contaminant
velocity (v
c
, cm/hr):
The pore-water velocity is operationally defined as the velocity of the nonadsorbing tracer.
3.12
R
f
'
n
n
e
%
K
d
D
b
n
e
(3.8)
R
f
' 1 %
K
d
D
b
n
e
(3.9)
R
f
' 1 %
K
d
D
b
n
(3.10)
R
f
' 1 %
K
d
D
b
2
(3.11)
The K
d
value can be calculated directly from the retardation factor (R
f
) and soil properties.
Depending upon the environmental conditions in which the contaminant moves and interacts with
the soil, the retardation factor can be correlated to the partition coefficient in a number of
different ways. At least 4 formulations of the retardation factor have been proposed [see reviews
in Bouwer (1991) and Whelan et al. (1987, 1996)]. These include the following:
where n = total porosity (cm
3
pore/cm
3
total volume),
n
e
= effective porosity (cm
3
pore/cm
3
total volume),
2 = volumetric water content in the vadose zone (cm
3
water/cm
3
total volume), and
D
b
= bulk density (g soil/cm
3
total volume).
Total porosity is the ratio of the air/water volume to the total soil. The effective porosity differs
from the total porosity in that the numerator is the volume of only those pore spaces that water
can travel through, excluding such void volumes as exist within aggregates or dead-end pore
spaces. Equation 3.10 was the original equation relating K
d
to R
f
. It was developed on an
empirical basis for use in chemical engineering and was first applied to groundwater situations by
Higgins (1959) and Baetslé (1967). Equations 3.8-3.11 were derived from the general transport
equation, which is the differential equation describing solute concentration changes in relation to
time, distance, dispersion coefficient, water velocity, soil bulk density, porosity, mass of solute per
unit dry mass of soil, and degradation of solute (Bouwer, 1991).
Equation 3.8 assumes that the soil has 2 types of pore spaces, those that permit flow to occur (n
e
),
and those pore spaces that do not permit flow to occur (n - n
e
). The contaminant in Equation 3.8
is assumed to migrate through the interconnected pore spaces, diffuse into dead-end pore spaces,
and instantaneously adsorb to or desorb from the soil matrix where fluid is and is not flowing.
Equation 3.8 also assumes that the solute concentration in the dead-end pore spaces is equivalent
to the solute concentration in the free-flowing spaces. Equation 3.8 has the appearance of being
more comprehensive than the other equations, but it does not allow the contaminant to travel with
the same speed as the fluid (i.e., nonadsorbing case), unless the total and effective porosities are
equal. Experience has shown that Equation 3.8 does not adequately reflect real-world
phenomena, suggesting deficiencies in our understanding of the geohydrochemical processes
3.13
impacting contaminant movement in the subsurface environment (Whelan et al., 1987).
Furthermore, in field studies, total porosity (n) can be measured directly, whereas effective
porosity (n
e
) can only be calculated from equations based on assumptions that are difficult to
defend (Freeze and Cherry, 1979).
Equation 3.9 includes the same processes as Equation 3.8, except that the contaminant does not
diffuse into the dead-end pore spaces. Many models use Equation 3.9 in their formulations. van
Genuchten and Wierenga (1986) suggest the use of Equation 3.10. Equation 3.10 includes the
same phenomena as Equation 3.9 except that the porous medium contains no dead-end pore
spaces. Again, the merit of its use over Equation 3.9 is that the measurable parameter n is
included in the formulation and not the calculated parameter n
e
. Equation 3.8, 3.9, and 3.10
describe chemical retardation in the saturated zone, whereas Equation 3.11 describes chemical
retardation in the unsaturated, or vadose, zone. As with Equation 3.10, contaminant transport
and retardation in Equation 3.11 occurs only in the free-flowing pore space. Bouwer (1991)
promotes the use of Equation 3.11 but defines the 2 term more generally as the water that is
moving, whether in unsaturated or saturated conditions. In his derivation of Equation 3.11,
Bouwer suggests that the 2 term also be used to quantify the mobile phases of water. Along with
van Genuchten and Wierenga (1986), he also contends that the use of Equation 3.11 allows better
distinction between retardation effects due to sorption and acceleration effects due to preferential
flow or anion exclusion.
Flow-through column experiments are appealing in that they allow observation of contaminant
migration rates in the presence of hydrodynamic effects (e.g., dispersion, colloidal transport, etc.),
and chemical phenomena (e.g., multiple species, reversibility, etc.). Ideally, flow-through column
experiments would be used exclusively for determining K
d
values, but equipment costs, time
constraints, experimental complexity, and data reduction uncertainties discourage widespread use.
One common problem in using column studies to measure K
d
values is that the breakthrough
curves are asymmetric. Such curves cannot be interpreted using Equations 3.8, 3.9, 3.10, or 3.11.
They require more complicated equations for solving for K
d
(Brusseau and Rao, 1989; van
Genuchten and Alves, 1982; van Genuchten and Wierenga, 1986).
One of the unique characteristics of measuring K
d
values from column experiments is that
nonequilibium conditions can be imposed. Especially under conditions in which the solute has
slow adsorption kinetics [e.g., those that may occur with uranium (Sposito, 1994)] or when
groundwater flow is fast, a measure of adsorption at equilibrium may over-estimate the extent to
which sorption occurs under actual field conditions. When either of these conditions are known
to exist in a study site, researchers should conduct column experiments at the flow rate existing in
the field, thereby creating realistic conditions.
Relyea (1982) provided an excellent review on the theoretical and experimental application of the
laboratory flow-through method of determining K
d
values. He reported that retardation factors
measured in column experiments depended on the water velocity and column dimension. For
short columns and slow water velocities, diffusion can become a major transport mechanism

3.14
v
p
'
v
d
n
e
(3.12)
resulting in lower retardation factors and lower K
d
values. At high velocities the effective pore
volume of a sample can decrease for short columns. High water velocities can also result in lower
retardation factors as a result of the solute not having sufficient time to adsorb to the soil, i.e.,
chemical equilibrium was not obtained. The effects of column length, mass of solute added to
column, diameter ratio of particle to column, and ratio of column diameter to column width on
the measured K
d
value were also presented by Relyea (1982).
3.2.4 Field Modeling Method
Field studies can provide accurate indications of the time of travel of the contaminant because the
concentrations of a dissolved contaminant are measured directly from samples taken from
monitoring wells. The field modeling method of estimating a K
d
value, also called the field
calibration method, uses a transport model and existing groundwater monitoring data. This
process, which is referred to as calibrating a groundwater transport model to K
d
values, involves
treating the K
d
value as an adjustable parameter (or dependent variable) while simulating
contaminant concentrations determined at monitoring wells. Groundwater calibration captures
the essence of the problem in the field. This is an iterative process that frequently requires the
adjusting the values for several other input parameters, such as effective porosity, dispersion, and
flow rate, to yield meaningful K
d
values. The minimum information that is needed for such a
calculation is the contaminant concentration at the source term, date of release, groundwater flow
path, groundwater flow rate, contaminant concentration at a monitoring well, distance between
source-release and monitoring well, dispersion coefficient, and source term. The retardation of
the chemical is then estimated as the ratio of the pore-water velocity to the contaminant velocity
(Equation 3.7). The pore-water velocity, v
p
, can be based on Darcy’s law (Freeze and Cherry,
1979) where
where v
d
= Darcy velocity
n
e
= effective porosity
However, 2 key drawbacks to this technique is that it is highly site specific and very model
dependent. Additionally, many assumptions have to be made about the water flow in the study
site including uniform flow and flow path. Not obvious, is that the K
d
value calculated by this
method greatly improves with more data. A detailed description of the theory of calculating K
d
values by this method and some examples of this approach are presented in Chapter 4.
3.2.5 K
oc
Method
The extent to which an organic contaminant partitions between the solid and solution phases is
determined by several physical and chemical properties of the contaminant and soil (Lyman et al.,
1982). Since most sorption of hydrophobic organic substances is to the natural organic matter
present in sediments or soils, the usual approach is to assume that all sorption is to that matter and

1
Other limits have been suggested for the minimum organic fraction, f
oc
, such as less than
0.1 percent (EPA, 1989) or less than a few tenths of a percent (Pignatello, 1989).
3.15
K
d
' K
oc
f
oc
(3.13)
to invoke a partition coefficient between organic carbon (K
oc
) or organic matter (K
om
) and water
(Seth et al., 1999). Hydrophobic solutes appear to bind readily and rapidly with the outer surface
region in a few hours to a few days and then diffuse slowly into (and out of) the hydrophobic
interior region and narrow cavities in the sediment or soil organic matter during time periods of
weeks (Seth et al., 1999). An empirical approach that has had wide acceptance in the scientific
community is the organic-carbon partitioning coefficient (K
oc
) method introduced by Karickoff et
al. (1979).
For this method, sorption of an organic contaminant, such as polynuclear aromatic hydrocarbon
(PAH), is assumed to occur only to the organic material in the soil. The partitioning between the
solid and solution phases is expressed as:
where K
oc
= ratio of the contaminant concentration on the organic matter on a dry weight basis
to its dissolved concentration in the surrounding fluid (ml/g) and
f
oc
= fraction of organic carbon in the soil (mg/mg).
Importantly, the K
oc
method is only applicable for estimating organic compound partitioning.
Gschwend and Wu (1985) report that if precautions are taken to eliminate or account for
nonsettling microparticles or organic macromolecules which remain in the aqueous phase during
laboratory sorption tests, the observed organic-carbon partitioning coefficient have been found to
remain constant over a wide range of environmental and experimental conditions. However,
recent studies by Chiou et al. (1998) and Seth et al. (1999) indicate that for any given chemical,
an inherent variability in K
oc
values is expected as a result of different environmental conditions
and equilibrium times. Dragun (1988) identified the following conditions when this approach is
less accurate:
C When the organic fraction, f
oc
, is less than 1.0 percent
1
(LaGrega, 1994) or greater than
20 percent (EPA, 1988)
C When there are large amounts of swelling clays present (e.g., montmorillonite)
C When the partitioning organic compound is polar
C When mechanisms other than simple partitioning contribute to adsorption (e.g., cation-
exchange, anion-exchange)
C When a substantial time is required to reach equilibrium
The organic content of most soils falls in the range of 0.2 to 3.0% (LaGrega, 1994).
3.16
K
oc
' "K
ow
(3.14)
K
d
' 10
&4
K
oc
[57.735(C
om
) % 2.0(C
clay
) % 0.4(C
silt
) % 0.005(C
sand
)] .
(3.15)
The commonest correlation for K
oc
is with the octanol-water partition coefficient (K
ow
) for which
extensive databases and reliable estimation methods exist (Seth et al., 1999). A simplified
relationship between these two parameter is given by Equation 3.14.
where " = correlation coefficent (unitless).
LaGrega (1994) reports a value of (" = 0.63) as a commonly used value while Seth et al. (1999)
calculate a value of (" = 0.35) with a variation in " by a factor of 2.5 in either direction. Seth et
al. (1999) also suggested that K
oc
estimates be viewed as a distribution, which includes
uncertainties about attainment of equilibrium and the variability in the composition of organic
matter present in soils and sediments, rather than as a single point value.
Strenge and Peterson (1989) applied the principle of the K
oc
model to estimating the partition
coefficients for organic compounds on soils. They defined the K
d
for organic compounds through
the combination of the K
oc
model and a parametric model (discussed in Section 3.4.2) based on
the concentrations of organic material (C
om
, percent w/w), clay (C
clay
, percent w/w), silt (C
silt
,
percent w/w) and sand (C
sand
, percent w/w) as dependent variables:
Equation 3.15 has the disadvantage of requiring more input parameters than Equations 3.13 and
3.14, but it provides an innovative approach for estimating the K
d
of organic compounds.
3.3 Issues Regarding Measuring and Selecting K
d
Values
3.3.1 Using Simple Versus Complex Systems to Measure K
d
Values
Soils are a complex mixture of solid, gaseous, and liquid phases. Each phase contains several
different constituents. Sposito (1989) estimated that the aqueous phase of a typical soil easily
contains between 100 and 200 different soluble complexes, many of them involving metal cations
and organic ligands (Table 3.1). The main effect of pH on these complexes, as is evident in
Table 3.1, is to favor free metal cations and protonated anions at low pH and carbonate or
hydroxyl complexes at high pH. The number of soluble complexes are also likely to be greater in
systems with elevated pH and organic matter concentrations. The solid phase in natural soils
typically contains more than 10 different constituents, including minerals, microbes, oxides,
naturally occurring organic matter, and organic, carbonate and/or oxide (e.g., iron, aluminum, and
manganese) coatings. The gas phase is quite different from that of above ground air as a result of
its interaction with the other phases and effects of pressure, temperatures, and microbial activity
(Sposito, 1989). For instance, the carbon dioxide levels is commonly several orders of magnitude
greater in soils than in above ground air (Wood and Petratis, 1984).

3.17
Table 3.1. Representative chemical species in acidic and basic soil solutions (after Sposito,
1989).
Cation
Principal Species
1
Acid Soils Alkaline Soils
Aluminum Al-org,
2
AlF
2+
, AlOH
2+
Al(OH)
4
-
, Al-org
Cadmium Cd
2+
, CdSO
4
E
(aq), CdCl
+
Cd
2+
, CdCl
+
, Cd SO
4
E
(aq), CdHCO
3
+
Calcium Ca
2+
, CaSO
4
E
(aq), Ca-org Ca
2+
, Ca SO
4
E
(aq), CaHCO
3
+
Chromium(III) CrOH
2+
Cr(OH)
4
-
Chromium(VI) CrO
4
2-
CrO
4
2-
Copper(II) Cu-org, Cu
2+
CuCO
3
E
(aq), Cu-org, CuB(OH)
4
+
,
Cu[B(OH)
4
]
4
E
(aq)
Iron(II) Fe
2+
, FeSO
4
E
(aq), FeH
2
PO
4
+
FeCO
3
E
(aq), Fe
2+
, FeHCO
3
+
, FeSO
4
E
(aq)
Iron(III) FeOH
2+
, Fe(OH)
3
E
(aq), Fe-org Fe(OH)
3
E
(aq), Fe-org
Lead Pb
2+
, Pb-org, PbSO
4
E
(aq),
PbHCO
3
+
Pb
2+
, PbHCO
3
+
, Pb-org, Pb(CO
3
)
2
2-
,
PbOH
+
Magnesium Mg
2+
, MgSO
4
E
(aq), Mg-org Mg
2+
, MgSO
4
E
(aq), MgCO
3
E
(aq)
Manganese(II) Mn
2+
, MnSO
4
E
(aq), Mn-org Mn
2+
, MnSO
4
E
(aq), MnCO
3
E
(aq),
MnHCO
3
+
, MnB(OH)
4
+
Molybdenum(VI) H
2
MoO
4
E
(aq), HMoO
4
-
HMoO
4
-
, MoO
4
2-
Nickel Ni
2+
, NiSO
4
E
(aq), NiHCO
3
+
,
Ni-org
NiCO
3
E
(aq), NiHCO
3
+
, Ni
2+
, NiB(OH)
4
+
Potassium K
+
K
+
, KSO
4
-
Silicon H
4
SiO
4
E
(aq) H
4
SiO
4
E
(aq)
Sodium Na
+
Na
+
, NaHCO
3
E
(aq), NaSO
4
-
Zinc Zn
2+
, ZnSO
4
E
(aq), Zn-org ZnHCO
3
+
, ZnCO
3
E
(aq), Zn-org, Zn
2+
,
ZnSO
4
E
(aq), ZnB(OH)
4
+
1
Complexes for each cation are listed in the order of their relative concentrations from
greatest to lowest concentration.
2
Org / Organic complexes (e.g., fulvic acid complexes).
3.18
Scientists will conduct geochemical studies with pure phases, such as goethite, quartz, or
montmorillonite to work in well-defined systems. They may also choose not to work with actual
groundwater, but instead work with a “synthesized groundwater,” such as a calcium chloride
solution or a calcium chloride/sodium chloride solution. Again, the intent is to work in a
chemically well-defined system with as few constituents as possible. Experiments conducted
under simplified systems have provided information about the mechanisms by which solutes
interact with solid surfaces (Sposito, 1984; Sposito, 1989), information that otherwise would not
be possible to obtain from experiments conducted with natural heterogeneous soils and ground-
water.
Ideally, for site-specific calculations, the transport modeler should use sorption values determined
for site-specific materials at site-specific conditions. In the absence of such data, the modeler
often selects a K
d
value taken from the literature that was measured under similar conditions as
existing at the study site. However, as discussed in Chapter 2 (Section 2.2.3), very subtle
properties of the solid and aqueous phases can have a profound affect on a contaminants K
d
. For
example, only 1 percent (w/w) organic matter existing as surface coatings in a South Carolina
surface soil completely masked the surface properties of the underlying minerals (Kaplan et al.,
1993). The organic coatings imposed a much greater sorption potential than would have been
expected based on mineralogical considerations. Similarly, the surface properties of the soils just
below these soils were entirely dominated by iron-oxide coatings (Seaman et al., 1995). The
effect of the iron-oxide coatings was to create a solid phase that was dominated by pH dependent
charge surfaces. These subsurface soils adsorbed large amounts of anions because the pH was
below the zero-point-of-charge [discussed in Chapter 2 (Section 2.2.3)] of the iron oxides, pH ~8.
Subtle changes in the aqueous composition in a batch K
d
test may also have a profound affect on
the measured K
d
value (Delegard and Barney, 1983). Thus, it is essential for the modeler
selecting K
d
values to recognize which solid and aqueous phase components have a strong affect
on the sorption of the contaminant of interest. Identifying these important components is the
subject of Volume II.
3.3.2 Field Variability
The purpose of any soil sample is to obtain information about a particular soil. The sample itself
is seldom, if ever, the entire soil mass in which one is interested. In statistics, this larger aggregate
of material, in which we are ultimately interested, is called the “population.” Information from the
sample is of interest only insofar as it yields information about the population, and the information
may or may not be representative, depending on how the sample is selected.
The population itself may be large or small, or even a part of what the modeler considers a larger
population. For contaminant transport modeling, the population is commonly defined by either
stratigraphic units or soil texture. The justification supporting the use of these definitions is based
both on practical and scientific considerations. Soil texture and stratigraphy can be easily and
inexpensively determined from well-log data and the close correlation of a number of hydrological
and chemical properties with soil textures is well documented (Petersen et al., 1996). A some-

3.19
what better definition of soil populations would be the cation- or anion-exchange capacity of the
soils. However, this option is appreciably more expensive and is valuable only for defining K
d
populations. The soil texture data are also used for defining water flow populations.
The intensity with which a soil must be sampled to estimate with given accuracy some
characteristic, such as K
d
value, will depend on the magnitude of the variation within the soil
population under consideration. The more heterogeneous the population, the more intense must
be the sampling rate to attain a given precision. In general, although differences have been found
to exist among lithographic units, considerable variation may be expected within the units for such
characteristics as pH, phosphorous, potassium, sodium, conductivity, volume weight, permeability
and porosity (Peterson and Calvin, 1986). In some instances, the variation within contiguous
units is so great that it is not feasible to estimate differences between the units with any
satisfactory degree of precision. For most characteristics, the variation, both within and among
units, decreases, with increasing depth in the profile (Peterson and Calvin, 1986). Hence,
subsurface environments generally need to be sampled less than surface soils to attain comparable
accuracy (Mackay et al., 1986; Warrick and Nielsen, 1980). Mackay et al. (1986) reported that a
number of soil properties, including K
d
values, changed more vertically than laterally.
3.3.3 The “Gravel Issue”
Because most K
d
values are measured in laboratory studies, the sample size has an upper mass
limit of about 100 g soil (and often 10 g with the increased emphasis of waste minimization and
high disposal costs for laboratory wastes) in batch K
d
measurements and several kilograms of soil
in column studies. Both tests also have particle size limitations. The batch K
d
is typically limited
to the less than 2-mm size fraction (Appendix C, ASTM, 1987; EPA, 1991; Roy et al., 1991).
This size fraction was selected for a number of reasons that are both practical and scientific in
nature. The less than 2-mm fraction has historically been defined as the soil fraction and the
greater than 2-mm fraction as the rock fraction. The less than 2-mm fraction is also convenient
for most standard glassware used in batch K
d
tests (Figure 3.1). Another practical consideration
is that greater uniformity of the soil sample and therefore of the measured K
d
value can be
achieved if the range of particle sizes used in the test is limited. Finally, the smallest fraction is the
most chemically reactive fraction due to its high specific surface area (m
2
/g). The particle size
used in column studies is also commonly limited to the less than 2-mm fraction. This size fraction
was selected for similar reasons as for the batch studies and to compare results between the 2
common methods. However, Relyea (1982) indicated that the less than 2-mm size fraction should
only be used in columns greater than 80 mm in diameter. He indicates that to avoid local velocity
effects (e.g., channeling or a radial velocity gradient), the column diameter should be at least 30 to
40 times the particle diameter of the solids used to pack the column.
The “gravel issue” is the problem that transport modelers face when converting laboratory-
derived K
d
values based on experiments conducted with the less than 2-mm fraction into values
that can be used in systems containing particles greater than 2 mm in size. As mentioned above,
the less than 2-mm fraction is the more chemically reactive fraction due primarily to its large

3.20
µ g metal
g sediment
µg metal
ml solution
'
ml
g
,
(3.16)
g metal
m
2
sediment
µg metal
ml solution
'
ml
m
2
,
(3.17)
surface area. There are many subsurface soils dominated by cobbles, gravel, or boulders. To base
the K
d
values on the less than 2-mm size fraction, which may constitute less than 1 percent of the
soil volume, would grossly overestimate the actual K
d
of the aquifer. Including large soil particles
in a K
d
determination will increase the cost of laboratory equipment and perhaps more importantly
will result in K
d
values with large error terms because of the great variability of the particle size
distribution in subsamples of a single soil.
Two general approaches have been proposed to address the “gravel issue.” The first is to assume
that all particles greater than 2 mm have a K
d
= 0 ml/g. As an example, if 75 percent (w/w) of a
formation is composed of particles greater than 2 mm and the K
d
value of the less than 2-mm size
fraction was 100 ml/g, then the K
d
value used in the model would be 25 ml/g. Although the
assumption underlying this approach is incorrect, the extent to which sorption occurs on these
larger particles may be small. This approach is likely to yield a more accurate value in systems
dominated by cobbles, gravel, and boulders.
The second approach is to normalize laboratory-derived K
d
values by surface area. Thus, instead
of having units of
the laboratory K
d
value would have units of
(Kaplan et al., 1995b). Theoretically, this latter approach is more satisfying because it permits
some sorption to occur on the >2-mm fraction and the extent of the sorption is proportional to the
surface area. The underlying assumption in this approach is that the mineralogy is similar in the
less than 2- and greater than 2-mm fractions and that the sorption processes occurring in the
smaller fraction are similar to those that occur in the larger fraction. Because sorption is a surface
area phenomena (Equation 3.16), as opposed to a weight phenomena (Equation 3.17),
normalizing the data to surface area has logic, and is commonly done is soil (Sposito, 1989) and
colloid chemistry (Alberty, 1987). The drawback to this approach is that an additional
measurement is needed to calculate the newly defined K
d
value in the laboratory. Specific surface
area measurement is a rather common and simple procedure (Carter et al., 1986). This approach
3.21
to the “gravel issue” also requires a means to convert available soil texture data, which are often
available from well-hole logs or outcroppings of the formation, into surface area data.
3.3.4 The “Colloid Issue”
The “colloid issue,” as it pertains to measuring K
d
values, is the problem experimentalists have in
separating the aqueous from the solid phases during a laboratory batch K
d
measurement (see
Chapter 2). Typically centrifugation or filtration are used to accomplish this. If contaminants are
sorbed to tiny particles that remain in suspension after the separation step, the experimenter will
incorrectly assign the sorbed contaminant to the dissolved phase, C
i
(Equation 3.2). This will
result in underestimating the true K
d
value. This is an especially important problem for
contaminants that sorb strongly to solids, especially organic matter. Organic matter has a much
lower density than clay (i.e., ~1.05 g/cm
3
for organic matter versus ~2.6 g/cm
3
clays) and
therefore the common centrifugation protocol may not be sufficient to separate the phases. Also,
organic matter may exist as extremely small particles, or molecules, ~0.005 µm in diameter.
Thus, when a great deal of organic matter is present in a soil, or when only a trace amount of
organic matter has a profound affect on the measured K
d
, additional precautions must be followed
(Gschwend and Wu, 1985). For example, Gschwend and Wu (1985) reported that they were able
to increase the partitioning coefficient, which is related to the K
d
term (see Section 3.2.3), of
polychlorinated biphenyl by 3 orders of magnitude by very carefully removing unsettled organic
particles from suspension.
Using a centrifuge to make the solid and solution phase separations can also result in the
formation of a very thin zone at the liquid surface where surface tension holds fine-grained
particles at the top of the solution. The experimentalist should look for such problems and avoid
sampling the surface of the clarified liquid. When pipets are used to remove supernatant solution,
the pipet should be inserted sufficient distance below the surface to avoid drawing in suspended
particles, but to a distance where the pipet tip is above the settled solids-liquid interface to avoid
drawing in previously settled fines.
For these reasons, many experimentalists prefer to filter the supernatant solution after
centrifugation. Filtration does have its problems, however. Filtering a small volume of
supernatant solution can bias the contaminant’s concentration if the filter membrane adsorbs
solute species. The type of filter membrane used effects the potential for adsorption. In our
experience polyethylene or other plastic-based filter membranes are more inert than cellulosic-
based membranes. Filter membranes can also be “pre-treated” with the supernatant solution by
discarding the first aliquot and filtering a second aliquot that is saved for analysis. If the filter
membrane does adsorb the analyte of interest, the amount adsorbed usually rapidly diminishes as
the volume of solution filtered increases. Thus discarding the first aliquot of filtered solution and
using subsequent aliquots for analysis lowers the chances of biasing final K
d
values.
Another problem associated with colloids, is that the traditional 2-phase solute transport model
does not account for contaminants moving in association with mobile colloids. This subject is
3.22
discussed in Chapter 2 (Section 2.5.1). Briefly, contaminants with a high affinity for sorbing to
rock or vadose zone soils are assumed to be retarded relative to the rate of groundwater flow.
However, an increasing body of evidence indicates that under some subsurface conditions,
components of the solid phase may exist as colloids that may be transported with the flowing
water. Association of contaminants with this additional mobile phase may enhance not only the
amount of the contaminant that is transported, but also the rate of contaminant transport. Most
current approaches to predicting contaminant transport ignore this mechanism not because it is
obscure or the mathematical algorithms have not been developed (Corapcioglu and Kim, 1995;
Mills et al., 1991), but because little information is available on the occurrence, the mineralogical
properties, the physicochemical properties, or the conditions conducive to the generation of
mobile colloids. There have been numerous examples in which mobile colloids have been
implicated as the vector responsible for enhanced transport (Kaplan et al., 1994a,b; Kaplan et al.,
1995a; reviewed by McCarthy and Degueldre, 1993).
3.3.5 Particle Concentration Effect
Many investigators have observed that K
d
values determined in the laboratory often exhibit a
dependence with respect to the ratio of solid to solution used in the measurements. As recently
discussed by Oscarson and Hume (1998), this dependence is puzzling. From a theoretical
perspective, the K
d
value should not depend on the solid-to-solution ratio, because the definition
of the K
d
model (see Equation 2.20) normalizes the ratio of the solute sorbed to the solid to the
solute concentration left in solution based on the mass of solid and solution used for the
measurement. Thus the K
d
has units of volume/mass, such as typically ml/g.
Investigators have often found that K
d
values measured for many contaminants for a given soil-
groundwater system decrease as the solid-to-solution ratio increases. For example, this particle
concentration effect on K
d
values has been observed by O’Conner and Connolly (1980), Oscarson
and Hume (1998), Honeyman and Santschi (1988), Meier et al. (1987), and others. The same
trend with respect to particle concentration has also been observed for K
d
values for organic
contaminants [e.g., see Gschwend and Wu (1985) and Voice et al. (1983)].
Investigators have offered several explanations for the observed dependency. These explanations
can be categorized into two groups: (1) “real” physical/chemical processes, and (2) experimental
artifacts. One rationalization offered in the “real” category is that the particle concentration
effect is thought to be caused by particle-particle interactions. In systems with higher solids
content, these interactions are perhaps physically blocking some adsorption sites from the
adsorbing solutes and thus causing decreased adsorption, or creating electrostatic interferences
such that the electrical surface charges on the closely packed particles diminish attractions
between the adsorbing solutes and surfaces of individual grains. In terms of physical effects,
individual particles in a slurry having a high solid-to-solution ratio may have a greater tendency to
coagulate and flocculate into larger particles that have less available surface adsorption sites than
individual grains and thus can adsorb less adsorbate. This phenomenon is likely exacerbated by
diffusion processes that in short-term laboratory measurements, do not allow sufficient time for
3.23
the adsorbate to diffuse to the internal surface adsorption sites. The net effect is a K
d
measurement with a lower value when a high solid-to-solution ratio is present, because not
enough time was allocated for the water/soil system to reach a final equilibrium state. Thus there
are possible experimental artifacts even within the context of this “real” process explanation.
Plausible experimental artifacts also include less efficient separation of the solid phase from high
solids-content slurries, such that more colloidal size particles laden with adsorbate remain in the
solution phase and the associated adsorbate gets included in the analysis of the solution phase.
Complexing agents may desorb and/or dissolve from the solids, and in turn compete with the
adsorbate for the available sorption surface sites. Soluble organic carbon is a common example of
this process. Such effects increase when higher solid contents are used. Other artifacts include
changes in the aqueous system that are caused by mass transfer from the larger quantity of solids
but are not recorded during these measurements. Another consideration in the category of
possible experimental artifacts for the solid-to-solution effect is improper data reduction.
McKinley and Jenne (1991) suggest that the so-called particle concentration effect often goes
away when adsorption data are replotted as adsorption isotherms, where the mass of solute
adsorbed per mass of solid and the concentration of solute in the equilibrium solution are plotted
on the y- and x-axes, respectively.
The explanations for the particle concentration (solid-to-solution ratio) effect are numerous and
still rather perplexing [see summaries by Oscarson and Hume (1998) and Jenne (1998a)]. Jenne
(1998a,b) includes valuable discussion on this “solids concentration effect.” He also presents
some recommendations that should be followed when performing adsorption experiments and
identifies several key issues that should be addressed by future adsorption research. One practical
position that has been supported by EPRI (1991) is to conduct adsorption experiments as close as
possible to the conditions that exist at the site where contaminant mobility is being simulated and
assessed. In most cases, this recommendation would require that K
d
values determined by flow-
through column testing would be preferred over batch measurements conducted at low solid-to-
solution ratios.
As noted above, it has been suggested that K
d
values may decrease with increasing solid-to-
solution ratios. If this is a real effect, application of K
d
values based on a batch experiment
conducted with a solid-to-solution ratio significantly less than those that would exist in the field
would therefore overestimate the magnitude of contaminant sorption and underestimate the extent
of contaminant migration.
3.4 Methods of Acquiring K
d
Values from the Literature for Screening Calculations
3.4.1 K
d
Look-Up Table Approach: Issues Regarding Selection of K
d
Values
from the Literature
Clearly, the greatest limitation of using a K
d
value to calculate a retardation term (Equations 3.8,
3.9, 3.10, and 3.11) is that it describes solute partitioning between the aqueous and solid phases
3.24
for only 1 set of environmental conditions. K
d
values are known to vary greatly with only slight
changes in the composition of the solid and aqueous phases and these conditions often vary
greatly in 1 study site. For example, when the aqueous chemistry for a batch K
d
measurement
was varied, americium K
d
values in a Hanford sediment ranged from 0.2 to 53 ml/g, greater than a
200-fold difference (Delegard and Barney, 1983). Additional variability in the americium K
d
values were observed when slightly different Hanford sediments were used: 4.0 to 28.6 ml/g
(Delegard and Barney, 1983). Similarly, Sheppard et al. (1976) measured americium K
d
values
ranging from 125 to 43,500 ml/g using identical aqueous phases but different soils.
An alternative approach to a constant K
d
model is one in which the K
d
value varies as a function
of a select group of environmental conditions (Delegard and Barney, 1983; Routson and Serne,
1972; Strenge and Peterson, 1989). The easiest variable K
d
model to interface with a transport
code is one based on a look-up table. For look-up tables, separate K
d
values are assigned to a
matrix of discrete categories defined by chemically important environmental parameters (Strenge
and Peterson, 1989; Whelan et al., 1992). Strenge and Peterson (1989) used 9 categories defined
by soil pH and texture in the Multimedia Environmental Pollutant Assessment System (MEPAS)
code. The 3 soil texture classes were <10 percent, 10 to 30 percent, >30 percent clay/organic
matter/oxide content. The 3 pH classes were >9, 5 to 9, and <5. The 9 cells defined by the pH
and soil texture classes contained literature-derived K
d
values and where data was not available,
estimated values were included in the table. The inorganic contaminants in the K
d
look-up table
were actinium, aluminum, americium, antimony, arsenic, asbestos, barium, beryllium, borate,
cadmium, calcium hypochlorite, calcium oxide, carbon, cerium, chlorate, chromium (III),
chromium (VI), cobalt, copper, curium, europium, fluoride, hydrogen fluoride, iodine, iron,
krypton, lead, lead oxide, lithium hydroxide, lithium ion, magnesium, manganese, mercury,
molybdenum, neptunium, nickel, niobium, nitrate, nitric acid, nitrogen dioxide, palladium,
phosphate ion, phosphorus,, plutonium, polonium, potassium hydroxide, potassium ion,
protactinium, radium, ruthenium, samarium, selenium, silicate ion, silver, sodium ion, strontium,
sulfate, sulphur, thallium, thorium, tin, tritium, uranium, vanadium, yttrium, zinc compounds, zinc,
and zirconium.
For any literature-derived K
d
value, it is essential to clearly understand the selection criteria and
the logic used to estimate K
d
values not found in the literature. For instance, Strenge and
Peterson (1989) reported a wide range of literature K
d
values for several cells, typically greater
than 10-fold and sometimes greater than a 100-fold difference between minimum and maximum
values. The values included in the MEPAS look-up table were the minimum values found in the
literature. They justified this criteria because they wanted to build conservatism into the code.
Conservatism is traditional when addressing the extent of contaminant migration and associated
health effects, but may be erroneous if the modeling calculations are being used to address
remediation options, such as pump-and-treat remediation. Conservatism for remediation
calculations would tend to error on the side of under estimating the extent of contaminant
desorption that would occur in the aquifer once pump-and-treat of soil flushing treatments
commenced. Such an estimate would provide an upper limit to time, money, and work required
to extract a contaminant from a soil. This would be accomplished by selecting a K
d
from the
3.25
upper range of literature K
d
values. Thus, the K
d
values in MEPAS would not provide a conser-
vative estimate for clean-up efforts .
Other important issues regarding the use of literature-derived K
d
values are illustrated in
Table 3.2. In any K
d
look-up table, a small number of ancillary parameters must be selected to
define the cells. pH and soil texture were the ancillary parameters used in the MEPAS code.
These are excellent general categories for a large number of contaminants, however, they are of
only secondary importance to a large number of other contaminants. For example, the amount of
vermiculite, which is a 2:1 layer silicate mineral common in the United States, especially in the
west and mid-west, is arguable the single most important ancillary parameter affecting cesium
sorption (Douglas, 1989). Redox state is another example of an ancillary parameter that is
extremely important relative to affecting the removal from redox-sensitive contaminants solution
[this is actually a precipitation process and not an adsorption phenomena (Ames and Rai, 1978;
Rai and Zachara, 1984; Sposito, 1989)]. Some important redox sensitive contaminants include
arsenic, chromium, molybdenum, neptunium, plutonium, selenium, technetium, and uranium. The
K
d
values of uranium in the 9 MEPAS categories range from 0 to 500 ml/g (Table 3.2).
Table 3.2. Example of a K
d
(ml/g) look-up table for uranium, uranium(VI), and uranium(IV).
Material
pH
$$ 9 5 - 9 ## 5
Fines
1
(%)
<10 10-30 >30 <10 10-30 >30 <10 10-30 >30
U
2
0 5 50 0 50 500 0 5 50
U(IV)
3
200 500 1,000 100 250 500 20 30 50
U(VI)
3
0 1 2 1 2 5 2 5 20
1
Fines (%) = sum of percentages of clay, organic matter, and hydrous-oxide in soil
2
Reference: Strenge and Peterson (1989)
3
Authors’ opinion based on values reported in Ames and Rai (1978), Ames and
McGarrah (1980), Cloninger et al. (1980), Cloninger and Cole (1981), Serne and Relyea
(1981), and Rai and Zachara. (1984).
By including an additional ancillary parameter of oxidation state, appreciably greater accuracy can
be assigned to K
d
values. For U(VI), the 9 categories may be assigned K
d
values in the range of 0
to 20 ml/g, whereas, as for U(IV), the 9 categories may be assigned K
d
values in the range of 20
to 1,000 ml/g. In this example, oxidation state is obviously a more important ancillary parameter
than soil texture and in systems with pH values greater than 5, oxidation state is more important

1
Strenge and Peterson (1989) generated most of the values in the pH>9 categories by
multiplying the K
d
values from the pH 5 to 9 category by 0.1. A significant quantity of K
d
values
exist in the literature for the latter pH category. The “0.1 factor” was based on consistent, but
flawed logic that metal contaminants are less likely to sorb because their cationic valence
decreases by [M
a+
(OH
x
)]
a-x
. It is now known that hydrolysis species adsorb as well as or better
than free cations (M
a+
). Also many contaminants precipitate at higher pH values, giving the
appearance of increased K
d
values. There are a few exceptions in pH >9 systems: Zr(OH)
5
-
species that may not adsorb as well as Zr
4+
, and UO
2
(CO
3
)
2
2-
and UO
2
(CO
3
)
3
4-
species do not
adsorb as well as the UO
2
2+
and UO
2
OH
+
species. The CO
3
2-
activity increases as pH increases so
complexes get more important at elevated pH levels.
3.26
than pH. The reduced form of U, U(IV), has a much greater K
d
value than U(VI) because the
former is known to precipitate from solution. The rather low uranium K
d
values reported by
Strenge and Peterson (1989) are somewhat misleading in that they represent, as mentioned above,
minimum values identified in the literature. These values would be entirely inappropriate for
modeling U(IV) transport. Thus, an important point to this discussion is that no single set of
ancillary parameters, such as pH and soil texture, is universally appropriate for defining categories
in K
d
look-up tables for all contaminants. Instead, the ancillary parameters used in look-up tables
must be based on the unique chemical properties of each contaminant.
An apparent inconsistency in Table 3.2 is that the minimum values selected by Strenge and
Peterson (1989) for the uranium data are greater than those for U(VI). This inconsistency is not
due to differences in literature used to estimate these values. Instead it arises from differences in
how K
d
values are estimated for cells in which no data are available.
1
This illustrates another
important reason for clearly understanding the criteria and process used in selecting data
incorporated into a look-up table. Clearly, differences in the criteria and process used to select K
d
values can result in appreciable different values included in a look-up table; in this example, as
much as 3 orders of magnitude.
3.4.2 Parametric K
d
Approach
The parametric K
d
approach is similar to that of the K
d
look-up table approach in that it varies K
d
values used in a transport model as a function of important ancillary parameters. It differs from
the K
d
look-up table in that it uses a regression equation to define the K
d
values instead of using
discrete categories. The K
d
value in this model varies as a function of empirically derived
relationships with aqueous and solid phase independent parameters. Thus, it has the distinct
advantage over look-up tables of having a continuum of K
d
values.
Factorial design experiments are most often used to determine the systematic change resulting
from varying the independent variables (e.g., pH, soil texture, and redox status) on the dependent
variables (uranium K
d
) (Box and Behnken, 1960; Cochran and Cox, 1957; Davies, 1954; Plackett
and Burman 1946). Statistical methods commonly used to derive quantitative predictor equations
include standard linear or nonlinear regression (Snedecor and Cochran, 1967), stepwise regression
3.27
Log K
d
(Americium) ' 2.0 % 0.1[NaOH] & 26.8[HEDTA] % 153.4[HEDTA]
2
(3.18)
(Hollander and Wolfe, 1973), and adaptive-learning networks (Mucciardi et al., 1979, 1980). All
these techniques have been used to develop empirical relationships describing K
d
values in terms
of other variables (Delegard and Barney, 1983; Routson and Serne, 1972; Serne et al., 1973;
Routson et al., 1981).
The empirical predictor equations commonly take the form of a nonlinear polynomial expression.
For example, after evaluating solutions consisting of several sodium salts, organic chelates, and
acids, Delegard and Barney (1983) came up with the following expression for an americium K
d
value:
where HEDTA is N-(2-hydroxyethyl) ethylenediaminetetraacetic acid. Numerous salts were
found to have no significant effect on americium K
d
values and therefore were not included in the
expression. Delegard and Barney (1983) also evaluated higher exponential and logarithmic terms
and determined that these terms did not improve the predictive capabilities of the expression (i.e.,
the regression coefficients were not significant at P # 0.05).
It is critical that parametric K
d
equations, such as Equation 3.18, be used to calculate K
d
values
for systems within the range of the independent variables used to create the equation. In the case
of Equation 3.18, the range of independent variables used in generate the model were selected to
simulate a plume beneath the Hanford Site in Richland, Washington. Using Equation 3.18 to
generate americium K
d
values for a plume low in pH and Na concentrations would not be
appropriate.
These types of statistical relationships are devoid of causality and therefore provide no certain
information regarding the mechanism by which the contaminant partitioned to the solid phase,
whether it be by adsorption, absorption, or precipitation. For example, the statistical analyses
may suggest a very strong relationship between pH and the K
d
term, when the actual sorption
process may be controlled by iron oxide adsorption. Because pH and iron-oxide charge are
covarients, a statistical relationship may suggest that sorption is due to pH, when in fact,
suggesting that sorption is solely caused by pH.
The parametric K
d
model is used in the transport equation, the code must also keep track of the
current value of the independent variables (e.g., [NaOH] and [HEDTA] for the examples
described in Equation 3.18) at each point in space and time to continually update the
concentration of the independent variables affecting the K
d
value. Thus, the code must track
many more parameters, and some numerical solving techniques (e.g., closed-form analytical
solutions) can no longer be used to perform the integration necessary to solve for concentration.
Generally, computer codes that can accommodate the parametric K
d
model use a chemical
subroutine to update the K
d
value used to determine the R
f
, when called by the main transport
code. The added complexity in solving the transport equation with the parametric K
d
sorption
model and its empirical nature may be the reasons this approach has been used sparingly.
3.28
3.4.3 Mechanistic Adsorption Models
Mechanistic models explicitly accommodate for the dependency of K
d
values on contaminant con-
centration, competing ion concentration, variable surface charge on the adsorbent, and solute
species solution distribution. Incorporating mechanistic, or semi-mechanistic, concepts into
models is attempted because the models become more robust and, perhaps more importantly from
the standpoint of regulators and the public, scientifically defensible. The complexity of installing
these mechanistic adsorption models into existing transport codes is difficult to accomplish.
Additionally, these models also require a more intense and costly data collection effort than will
likely be available to the majority of contaminant transport modelers who are conducting
screening calculations. Descriptions of the state of this science, with references to excellent
review articles, are presented in Chapter 2 and 5. A review of the methodology associated with
the determination of the constants for use in these mechanistic models, however, is beyond the
scope of this project. A review of the mechanistic adsorption models contained in EPA’s
MINTEQA2 geochemical reaction code is also presented in Chapter 5.
3.5 Summary
The objective of this chapter is to describe methods used to measure K
d
values. The advantages
and disadvantages and the assumptions underlying each method were discussed, and are
summarized in Table 3.3. A number of issues regarding the selection of K
d
values from the
literature for screening calculations are also addressed in this chapter. Specific issues discussed
included the use of simple versus complex systems to measure K
d
values, field variability, the
“gravel issue,” and the “colloid issue.”
Clearly, the greatest limitation of using a K
d
value to calculate a retardation term is that it is only
applicable to a single set of environmental conditions. Consequently, researchers have generated
K
d
values that varies as a function of ancillary environmental parameters. They include the look-
up table K
d
, the parametric K
d
, and the mechanistic K
d
. Models generated for parametric K
d
values have typically been for rather limited environmental conditions. Mechanistic K
d
values are
limited to uniform solid and aqueous systems with little application to the heterogenous soils that
exist in the natural environment. The easiest and the most common variable K
d
model to interface
with a transport code is the look-up table. No single set of ancillary parameters, such as pH and
soil texture, is universally appropriate for defining categories in K
d
look-up tables. Instead, the
ancillary parameters must vary in accordance to the geochemistry of the contaminant. It is
essential that the modeler fully understand the criteria and process used for selecting the values
incorporated in such a table. Just as important is to understand the logic used to estimate K
d
values not found in the literature. Differences in the criteria and process used to select K
d
values
can result in appreciable different K
d
values.
It is incumbent upon the transport modeler to understand the strengths and weaknesses of the
different K
d
methods and perhaps more importantly the underlying assumption of the methods in
order to properly select K
d
values from the literature. The K
d
values reported in the literature for
3.29
any given contaminant may vary by as much as 6 orders of magnitude. An understanding of the
important geochemical processes and knowledge of the important ancillary parameters affecting
the sorption chemistry of the contaminant of interest is necessary for selecting appropriate K
d
value(s) for contaminant transport modeling.

3.30
Table 3.3. Advantages, disadvantages, and assumptions of different methods used to determine
K
d
and the assumptions in applying these K
d
values to contaminant transport models.
Methods for Determining K
d
Batch
1
In-Situ Field Batch
Flow-Through Field Modeling K
oc
2
Minimum Input Data
3
C M
sed
C C
i
C C
0
C V
w
C C
i
C A
i
(or q
i
)
C C
o
C n, 2, or n
e
C D
particle
4
C >10 C
effluent
data
points
C C
o
tracer
C Time
C C
release
C C
well
C Time
C Distance
C v
w
C n, 2, or n
e
C Diffusion or
dispersion
coefficients
C K
oc
C f
oc
Advantages
C Inexpensive
C Quick
C In-situ
measurements
C Equilibrium
conditions
C Aqueous and solid
phases are precisely
those of the
modeled system
C Can measure sorp-
tion at field flow
rates, i.e., at non-
steady state condi-
tions
C Can measure hydro-
dynamic effects
(e.g., dispersion,
colloidal transport,
etc.) on R
f
, and
subsequently incor-
porate into
K
d
value
C Can measure effects
of chemical
phenomena (e.g.,
multiple species,
reversibility, etc.) on
R
f
and K
d
values.
C Derived K
d
has the
precise geochemical
conditions and flow
conditions of the
study site
C Fairly accurate
indirect method
C Often can use look-
up tables to get K
oc
value
C f
oc
is an easy
measurement
C K
oc
can be
correlated with K
ow
which has been
measured for many
different chemicals

3.31
Table 3.3. Continued.
Methods for Determining K
d
Batch
1
In-Situ Field Batch
Flow-Through Field Modeling K
oc
2
Disadvantages
C Provides estimate of
chemical processes
at equilibrium; flow
conditions are not
always at
equilibrium
C Physics involved not
considered
C Better mixing in
batch than in nature
C Typically uses larger
ratio of solution/soil
than exist in nature
C Experiments
measure adsorption
instead of
desorption, the
dominant process in
transport; desorption
is typically much
slower than
adsorption
C Speciation of
different forms not
considered
C Expensive to collect
samples
C Commonly have
high detection limits
(undesirable) for
measuring
contaminant on
solid phase (A
i
, q
i
)
C Site-specific data
C Cannot
unequivocally
differentiate
between, adsorbed,
precipitated, and
structural
constituents
C Commonly flow-
through system is
not at equilibrium
and therefore results
cannot be applied to
other flow
conditions
C Directly measure R
f
,
then back out K
d
;
therefore must make
assumptions about
relation between K
d
and R
f
5
C Measured K
d
values
commonly vary with
water velocity and
column dimensions
C Requires relatively
expensive
equipment
C Requires a lot of
time
C Complex experiment
to conduct
C Data are commonly
not well behaved,
i.e., asymmetric or
peakless break-
through curves
C Can investigate
some secondary
processes affecting
contaminant
transport, such as
effects of
unsaturated flow,
colloid-facilitated
contaminant
transport, mobile vs
immobile water
phases
C K
d
is truly site
specific
C K
d
is transport model
specific
C Need to make many
assumptions about
the water flow
including uniform
flow, direction, and
path length that
affect the calculated
K
d
value
C Measure R
f
, then
back out K
d
; many
assumptions go into
relating K
d
to R
f
C May or may not be in
equilibrium,
therefore not a
thermodynamic K
d
C K
d
value greatly
improves with more
field data collected
C Calculations can be
quite involved
C For organic
compounds only
C More hydrophobic
the contaminant
compound, more
accurate the K
d
; vice
versa with
hydrophilic
compounds

3.32
Table 3.3. Continued.
Methods for Determining K
d
Batch
1
In-Situ Field Batch
Flow-Through Field Modeling K
oc
2
Assumptions in Calculating K
d
C Adsorption rate =
desorption rate
C Only 1 type of
surface adsorption
site, A
C Only 1 type of
aqueous dissolved
species, C
i
C A>>>A
i
C Activity of A
i
=1
C Equilibrium has
been achieved
during mixing
period
C No adsorbate on
suspended colloids
C No precipitation of
adsorbate due to C
0
concentration
exceeding solubility
C Same as for batch
K
d
C Measurement of
adsorbed
contaminant, A
i
(q
i
),
can differentiate
between adsorbed,
precipitated, and
structural
constituents
C Must assume a
relationship between
R
f
and K
d
5
C Water flow and
dispersion
coefficient is
constant
C Must assume a
relationship between
R
f
and K
d
C Know n
e
or n
C Must know the flow
path and velocity of
plume
C Sorption is uniform
in the study size
C Same as for
laboratory batch K
d
C Organic
contaminant sorbs
(partition) only to
organic matter in
soil, no sorption
occurs to inorganic
phases

3.33
Table 3.3. Continued.
Methods for Determining K
d
Batch
1
In-Situ Field Batch
Flow-Through Field Modeling K
oc
2
Assumptions in Applying Measured K
d
to Transport Model
C Adsorption of solute
is linear, i.e., it is
independent of C
i
C Adsorption of solute
is reversible
C Solute movement is
slow enough that
equilibrium
conditions exist
between the solute
and soil
C Geochemical
conditions (presence
and concentration of
background
electrolytes and
solid phases) of
batch experiment are
identical to those in
aquifer
C Temperature and
pressure conditions
of batch experiment
are identical to those
in the aquifer
C Mixing in aquifer is
as thorough as in
batch experiment
C Difference between
the soil/water ratio
in the aquifer and
batch experiment is
not important to K
d
value
C Same as for
laboratory batch K
d
(except fourth point
not relevant to
in-situ batch
method)
C It is more common
to enter the R
f
value
derived from
experiment than the
K
d
value into
transport code;
consequently, do not
need to make any
assumptions about
the relationship
between R
f
and K
d
C If R
f
value from
column experiment
is entered into
transport code and
the flow conditions
of the experiment
are similar to those
in the site being
modeled, then no
assumptions need to
be made regarding
affect of
nonequilibium
conditions
C None C Same as for
laboratory batch K
d
1
See Equation 3.4
2
See Equation 3.13
3
A = Concentration of free or unoccupied surface adsorption site on a solid phase; C
i
= the total dissolved adsorbate
concentration remaining in solution at equilibrium; f
oc
= fraction of soil that is organic carbon; C
release
= concentration of
solute at time of release; C
well
= concentration of solute in monitoring well; M
s
= soil mass; A
i
= the concentration of
adsorbate on the solid at equilibrium; n = total porosity; n
e
= effective porosity; V
W
= solution volume; D
b
= bulk density;
D
particle
= particle density; 2 = water saturation.
4
D
particle
is used to calculate bulk density (D
b
); D
b
= [D
particle
(1 - n)].
5
See Equations 3.8, 3.9, 3.10, and 3.11.

1
A list of acronyms, abbreviations, symbols, and notation is given in Appendix A. A list of
definitions is given in Appendix B
4.1
4.0 Groundwater Calibration Assessment Based on Partition
Coefficients: Derivation and Examples
4.1 Introduction
Partition (or distribution) coefficient, K
d
,
1
values are utilized in transport and risk assessment
modeling because of their simplicity in (1) understanding, (2) measuring, and (3) providing
closed-form, explicit, analytical solutions to the advective-dispersive equations. Whelan (1996)
presented a discussion that illustrates the inherent difficulties associated with utilizing partition
coefficients as the sole parameter to define the geochemical properties of a solute as it migrates
through a subsurface environment. Multiple definitions for K
d
values have been identified,
including those based on thermodynamics (e.g., Gibbs free energy of formation), experiments
(e.g., batch and flow-through tests), and theory (e.g., isotherms). Each of these procedures
identifies a different value for the same parameter, which is supposed to describe the same
phenomena. Although K
d
values can be thermodynamically defined, their meaning becomes less
clear in the real world. As such, K
d
values can be estimated using transport models. This process,
called calibrating a groundwater transport model to K
d
values, involves treating the K
d
value as
the adjustable parameter (or dependent variable) while simulating known monitored contaminant
data. Groundwater calibration captures the essence of the problem in the field. This is an
iterative process that frequently requires that the magnitude of a number of other input
parameters, such as effective porosity, dispersion, and flow rate, be adjusted to yield meaningful
K
d
values. A K
d
value represents one of the calibration parameters because its magnitude is
subject to not only the laboratory analyses but also to the heterogeneity in the field and different
ways it is used in the mathematical constructs of different models.
4.2 Calibration: Location, Arrival Time, and Concentration
When calibrating a groundwater model to monitored information (e.g., concentrations at a
monitoring well), the model must predict the correct arrival time at the correct location, matching
the magnitude of the monitored concentration. Therefore, time, location and magnitude are
3 crucial elements associated with any calibration exercise, and K
d
impacts two of them (i.e.,
travel time and magnitude). Location is predetermined by the user with respect to monitoring
wells, receptor locations, etc. Once the distances have been defined, the calibration requires
modifications to parameters that govern travel times and concentration levels. Parameters, which
influence water and contaminant movement, are varied within acceptable ranges in an attempt to
recreate conditions in the field. As the model complexity increases, the number of parameters that
the analyst can vary increases, and the calibration process becomes increasingly more
complicated. Figure 4.1 illustrates the relative relationships between input-data quality, output
uncertainty, and types of problems addressed by each level of assessment. As Figure 4.1 indi-
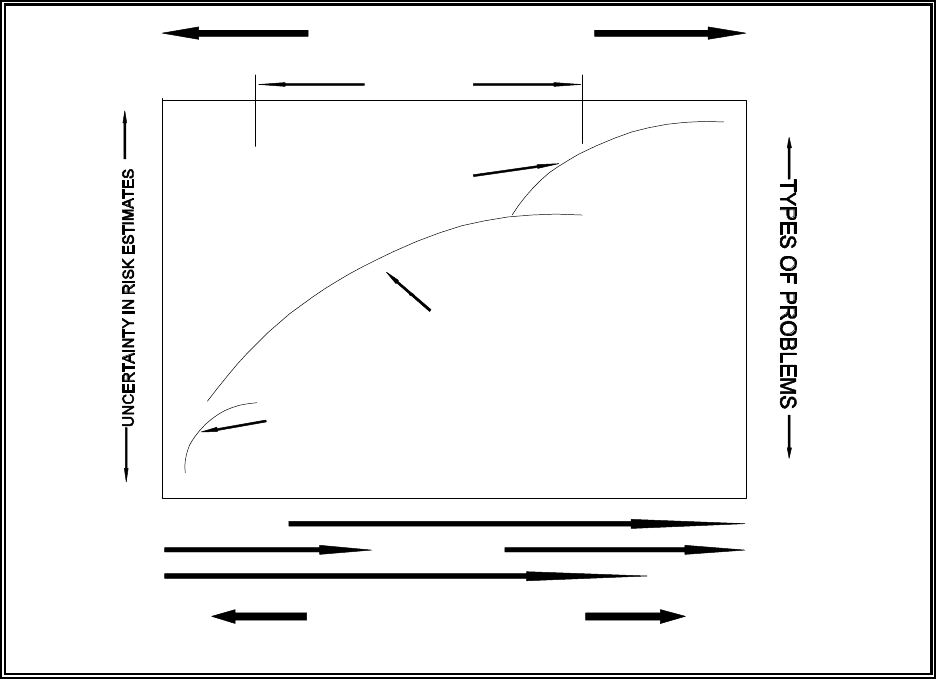
4.2
LEVEL OF ANALYSIS
specific
site-
broad
range
least
greatest
screening
analytical
numerical
analytical
screening
numerical
INPUT DATA QUALITY
site-specific
regional
representative
highestleast
cates, the computational requirements tend to be less at the earlier stages of an assessment when
available data are less, and, correspondingly, the uncertainty with the output results tends to be
greater. As the assessment progresses, improved site-characterization data and conceptualization
of the problem increase, thereby reducing the overall uncertainty in risk estimates.
Figure 4.1 also illustrates some of the characteristics and relationships between screening-level
(ranking), “analytical” (prioritization and preliminary assessments), and numerical (detailed)
models.
Figure 4.1. Relative relationships between input-data quality, output uncertainty, and types
of problems addressed by each level of assessment.
4.3
Screening models are used to identify environmental concerns. These models, often based on a
structured-value approach, are designed to be used with regional/representative information.
Models such as the Hazard Ranking System (HRS) (EPA, 1984, 1992b) divide site and release
characteristics into predetermined categories that are assigned a point value based on answers to
questions. The score from such systems is useful to determine if a situation requires further
analysis, but not to provide a method for estimating actual concentrations or impacts in the
environment.
Detailed analyzes require a highly specialized assessment of potential impacts. Detailed analyses
are usually reserved for the most complex models, are data intensive, and are based on the
expertise of the analyst. These detailed assessment models are used to address complex problems
and concerns that are relatively well-defined. Models for detailed analyses tend to focus on
special sets of problems and special types of situations. Although detailed assessment tools are
appropriate for their intended application, extension beyond the site-specific application is often
difficult or cost prohibitive. Typical models include MODFLOW (McDonald and Harbaugh,
1988) and CFEST (Cole et al., 1988).
Analytically/semianalytically/empirically based models (designated as “analytical” models in
Figure 4.1) can be utilized for prioritization or preliminary assessments and exist between initial-
screening and highly specialized numerical models. These physics-based models are the most
versatile as they do not have the data constraints associated with the numerical models. The
analytical models may contain some numerical computations, hence the semianalytical designa-
tion. As Figure 4.1 illustrates, the analytical models are designed to provide environmental
evaluations over a wide range of applications. Groundwater models that fall into this category
include AT123D (Yeh, 1981), GROUND and GRDFLX (Codell et al., 1982), and MEPAS (Buck
et al., 1995; Whelan et al., 1992). The analytical-assessment models are codes with physics-
based algorithms whose components can be utilized in a detailed (i.e., numerical) or an initial-
screening (i.e., ranking/prioritization) assessment, where data and circumstances warrant.
The calibration process is an interactive one. Because data tend to be limiting, there are generally
multiple ways in which parameters can be combined so the simulation results match monitored
information. With increasing number of monitored data available, less combinations of the
modeling parameters are possible to match the monitored information. In addition, many of these
“matches” can assign unrealistic values to parameters; therefore, the number of acceptable
possible combinations becomes even more limited. When calibrating, parameters can only be
varied within ranges that physically make sense for the site and its conditions. If unrealistic
output is a result of the analysis, then the (1) conceptual site model has to be re-evaluated, (2)
input data must be re-examined, and/or (3) model must be re-evaluated to ensure that the
assessment does not violate the assumptions, limitations, and constraints associated with the
mathematical constructs of the code.
Each code has its own mathematical equations upon which it is based. A calibration exercise is
performed to meet the constructs of these equations. Analytical models tend to be easier to work

4.4
MC
i
Mt
% v
(
M C
i
Mx
' D
(
x
M
2
C
i
Mx
2
% D
(
y
M
2
C
i
My
2
% D
(
z
M
2
C
i
M z
2
& 8 C
i
(4.1)
with because of their closed-form, explicit solutions. With an analytical model, some initial
calculations can be made that can provide an initial starting point for the calibration process; this
process also illustrates how retardation factors (and ultimately K
d
) influence the calibration
process. As noted earlier, the intent of the calibration process is to get a contaminant from a
source to the monitored location (e.g., monitoring well) at the proper time with the appropriate
concentration. In addition, the amount of mass monitored in the environment must be conserved,
that is, the amount of mass predicted by the model to be in the environment should match the
amount of mass monitored in the environment.
Travel times are influenced by the retardation factor, pore-water velocity, and dispersivity,
although other parameters can also influence the outcomes. The retardation factor can be directly
impacted by K
d
. In the vadose zone, soil type and moisture content influence pore-water velocity,
and in the saturated zone, soil type and effective porosity influence pore-water velocity.
Longitudinal dispersivity normally influences the time to peak but by no more than 10 percent,
although more is possible. Concentrations are generally influenced by the contaminant flux rate
(or total mass released into the environment), mixing distances (dilution), pore-water velocity
(dilution), retardation factor (K
d
), and dispersivity (dispersion). If the size of the source is not
well known, the areal extent of contamination influences concentration levels for spatially near-
field problems. In any modeling exercise, the analyst will know some of the general charac-
teristics of the parameters. Typically, the parameters that are used to calibrate the model are not
known exactly; therefore, they can be modified within an appropriate range to help the analyst
capture the essence of the problem.
4.3 Illustrative Calculations to Help Quantify K
d
Using Analytical Models
If K
d
forms the basic premise for retarding the movement of contaminants in a subsurface
environment in the mathematical algorithms of a groundwater transport code, then the K
d
permeates all of the contaminant transport calculations. Different computer codes may use
different mathematical constructs, but the influence of K
d
is usually very pronounced. The K
d
value influences the calculations for determining the (1) contaminant travel time, (2) mass of
contamination at the source or in a plume, and (3) distribution of the concentration in the
environment. As an illustration of the impact that the K
d
parameter can have in transport
calculations, the influences of K
d
on an analytical solution to the advective-dispersive equation are
explored.
4.3.1 Governing Equations
The 1-dimensional advective, 3-dimensional dispersive equation with first-order
degradation/decay can be expressed as follows:
4.5
v
(
'
v
p
R
f
(4.2)
v
p
'
v
d
n
e
(4.3)
R
f
' 1 %
D
b
K
d
n
e
saturated
(4.4)
R
f
' 1 %
D
b
K
d
2
vz
vadose
(4.5)
K
d
'
A
i
C
i
(4.6)
D
(
'
D
mech
% D
mol
R
f
(4.7)
D
mech
' " v
p
(4.8)
in which
where C
i
= dissolved concentration
v* = contaminant velocity
D* = dispersion coefficient in the x, y, and z directions adjusted for retardation with
the retardation factor
8 = first-order degradation/decay coefficient
v
p
= pore-water velocity
v
d
= Darcy velocity
R
f
= retardation factor
n
e
= effective porosity
D
b
= bulk density
2
vz
= moisture content in the vadose zone
K
d
= partition (distribution) coefficient
A
i
= adsorbed contaminant concentration on the soil particles
D
mech
= mechanical dispersion
D
mol
= molecular diffusion coefficient
4.6
C
i
' *' X
Gf
Y
Gf
Z
Gf
(4.9)
*' '
M
rel
R
f
n
e
(4.10)
X
Gf
'
1
4 B D
(
x
t
1/2
exp (&8 t) exp &
(x & v
(
p
t)
2
4 D
(
x
t
(4.11)
Y
Gf
'
1
4 B D
(
y
t
1/2
exp &
y
2
4 D
(
y
t
(4.12)
Z
Gf
'
1
h
m
(4.13)
" = dispersivity in the x, y, or z direction
The solution of advective-dispersive equation for an instantaneous release through a point source
in a saturated zone, which is uniformly mixed in the vertical direction, at a distance (x) down
gradient from the center of the source is as follows (Codell et al., 1982; Fischer et al., 1979;
Whelan et al., 1996; Yeh and Tsai, 1976; Yeh 1981):
where *' = mass-related constant
X
GF
, Y
Gf
, and Z
Gf
= Green's functions (which are orthogonal) in the x, y, and z
directions, respectively
X
GF
= Green’s function corresponding to flow direction
in which
where M
rel
= released mass
y = off-centerline distance
h
m
= mixing-zone thickness
and all other parameters retain their previous definitions.
The impact that the retardation factor and, hence, K
d
has on the calculated value of the
concentration at the receptor location can be profound, as illustrated by the number of locations
that these terms appear in the governing equations.
4.7
t
T
'
x
v
(
(4.14)
t
T
' t
vz
% t
sz
'
H
1
R
f1
v
p1
%
x
2
R
f2
v
p2
(4.15)
t
T
'
(H
1
) 1 %
D
b1
K
d1
2
vz
v
p1
%
(x
2
) 1 %
D
b2
K
d2
n
2
v
p2
(4.16)
> '
K
d1
K
d2
(4.17)
t
T
'
H
1
v
p1
1 %
D
b1
>
2
vz
K
d2
%
x
2
v
p2
1 %
D
b2
n
e2
K
d2
(4.18)
4.3.2 Travel Time and the Partition Coefficient
As previously noted, it is very important to ensure that the contaminant arrives at the monitoring
location at the appropriate time, and K
d
can have a profound impact on the travel time. The
advective travel time of the contaminant is defined as the distance x traveled divided by the
contaminant velocity:
where t
T
= total advective travel time of the contaminant
If a contaminant is traveling from a contaminated source through a vadose zone, through a
saturated zone to a monitoring location, the total advective travel time is the summation of the
travel times through the vadose (t
vz
) and saturated (v
sat
) zones:
where H
1
= thickness of the vadose zone
subscripts 1 and 2 = vadose and saturated zones, respectively
Substituting the definitions for retardation factor gives a slightly modified equation:
This equation demonstrates the potential impact that K
d
has on the travel time. Because K
d
is
assumed to be constant over the distanced traveled, a constant, >, can be defined, which
represents the ratio of the partition coefficients between the vadose and saturated zones:
Substituting > into the total travel time equation gives the travel time as a function of the saturated
zone’s partition coefficient:

4.8
t
T
'
H
1
v
p1
%
H
1
D
b1
>
v
p1
2
vz
%
x
2
D
b2
v
p2
n
e2
K
d2
%
x
2
v
p2
(4.19)
K
d2
'
t
T
& (H
1
/v
p1
) & (x
2
/v
p2
)
H
1
D
b1
>
v
p1
2
vz
%
x
2
D
b2
v
p2
n
e2
(4.20)
C
i
'
A
i
K
d
(4.21)
M
ads
' V
source
A
i
1 & n D
particle
(4.22)
M
aq
'
V
source
A
i
2
vz
K
d
(4.23)
Rearranging this equation and solving for K
d2
gives:
This equation can be used to estimate initial values for the partition coefficients in the vadose and
saturated zones, which will help ensure that the contaminant reaches the monitoring location at
the appropriate time. These values can also be compared to literature or experimental values to
see if they are consistent. If not, then the conceptual site model must be re-analyzed to ensure
that the proper problem has been captured or that the appropriate data are being utilized.
4.3.3 Mass and the Partition Coefficient
The partition coefficient can be used to help estimate the mass of contamination that exists at the
source or in a plume. The reported soil contamination in the vadose zone is usually expressed as
the adsorbed concentration (A
i
) and typically has units of mg/kg, which is also expressed as ppm
(parts per 10
6
). The aqueous concentration (C
i
), using K
d
as a conversion factor, can be
calculated as follows:
The mass associated with the adsorbed phase in the vadose zone can be estimated as:
where M
ads
= mass associated with the adsorbed phase in the vadose zone
V
source
= volume associated with the contaminated source
n = total porosity
D
particle
= particle density
The mass associated with the aqueous phase in the vadose zone can be estimated as:

4.9
M
vadose
' M
ads
% M
aq
vadose
(4.24)
M
aq
' V
source
C
i
n
(4.25)
M
ads
' V
source
C
i
K
d
1 & n D
particle
(4.26)
M
saturated
' M
ads
% M
aq
saturated
(4.27)
M
Total
' M
vadose
% M
saturated
(4.28)
C
i
'
C
Tp
D
b
2
vz
% D
b
K
d
(4.29)
A
i
'
C
Tp
D
b
K
d
2
vz
% D
b
K
d
(4.30)
where M
aq
= mass associated with the aqueous phase in the vadose zone
The total mass associated with the vadose zone represents the summation of the mass associated
with the adsorbed and aqueous phases, assuming no free product:
where M
vadose
= total mass associated with the vadose zone
The reported aqueous contamination in the saturated zone is usually expressed as the dissolved
concentration C
i
and typically has units of mg/l, which is also expressed as ppm (parts per 10
6
).
The mass associated with the aqueous phase in the saturated zone can be estimated as:
The mass associated with the adsorbed phase in the saturated zone can be estimated as:
The total mass in the vadose zone represents the summation of the mass associated with the
adsorbed and aqueous phases, assuming no free product:
where M
saturated
= total mass associated with the saturated zone
The total mass in the system is the summation of the masses in the vadose and saturated zones:
If the environmental contamination in the vadose zone is expressed as a total mass in the waste
site (or layer) per dry weight of soil, the dissolved and adsorbed concentrations can be calculated
as follows (Whelan et al., 1987):
where C
Tp
= total mass at the site per dry weight of soil
4.10
C
i
'
C
T
2
vz
% D
b
K
d
(4.31)
A
i
'
C
T
K
d
2
vz
% D
b
K
d
(4.32)
MC
i
Mt
' D
(
y
M
2
C
i
My
2
(4.33)
C
i
'
M
A
F
sd
(2 B)
1/2
exp &
y
2
2 F
2
sd
(4.34)
F
sd
' 2 D
(
y
t
1/2
'
2 D
y
t
R
f
1/2
(4.35)
D
y
' "
y
v
p
(4.36)
If the environmental contamination is expressed as a total mass per total volume of the waste site
(or soil layer), the dissolved and adsorbed concentrations can be calculated as follows (Whelan et
al., 1987):
where C
T
= total mass at the site per total site volume
4.3.4 Dispersion and the Partition Coefficient
The 1-dimensional, dispersive equation in the lateral direction can be expressed as
where all of the terms are as previously defined. For an instantaneous release from a unit area in
an aquifer of infinite lateral extent, the time-varying concentration as a function of lateral distance
off the center line can be expressed as follows:
in which
where M
A
= instantaneous mass released per unit area (i.e., instantaneous point-source release)
F
sd
= standard deviation associated with the Gaussian solution
Note that the standard deviation (i.e., the degree of lateral spreading) is a function of the
retardation factor and, hence, K
d
.

4.11
t '
x R
f
v
p
(4.37)
F
sd
' 2 "
y
x
1/2
(4.38)
To gain an understanding of the impact of the retardation factor (and K
d
) on simple advective-
dispersive systems, the impact of retardation at a location, x, can be discerned by substituting the
time, t, with the advective travel time, as follows:
The standard deviation that indicates the degree of spread at location x is independent of the
retardation factor and K
d
. This phenomenon is expected because when combined with flow in the
longitudinal direction, advection impacts the effects of dispersion in the lateral direction. In
effect, advection transports the contaminant in the longitudinal direction, so there is no infinite
dispersion at any location in the lateral direction. Hence, Gaussian plumes grow as they migrate
down gradient. Unlike the contaminant travel time, the dispersive phenomenon is not closely tied
to K
d
4.4 Modeling Sensitivities to Variations in the Partition Coefficient
Because the retardation factor and partition coefficient permeate many aspects associated with the
mathematical algorithms for contaminant transport in the subsurface environment, K
d
can have a
significant impact on the outcome of any modeling exercise. Under certain circumstances though,
K
d
can have very little impact on the outcome. The next 2 sections discuss the conditions under
which partition coefficients influence the outcome.
1. Relationship Between Partition Coefficients and Risk -- This section explores the
situations under which variations in K
d
can have a significant influence on simulated
groundwater concentrations and, hence, risk.
2. Partition Coefficient as a Calibration Parameter in Transport Modeling -- This section
presents an illustrative example of a calibration exercise where the calibration parameter is
the partition coefficient.
4.4.1 Relationship Between Partition Coefficients and Risk
The K
d
parameter potentially has a very large impact on the mobility of constituents in a
subsurface environment. When combined with other phenomena (e.g., degradation/decay,
dispersion, pore-water velocity), K
d
can have a significant impact by redistributing the
contaminant both spatially and temporally.
For example, when the K
d
parameter is sufficiently large, the contaminant moves slowly from the
source to the receptor. Because significant levels of contaminant have not reached the receptor

1
The MEPAS simulations were based on actual site data published in Lewis et al. (1994) for
the CERCLA hazardous waste site ST-19. Only the contaminant information was changed.
Radionuclides were never present at the site; they are only used here as an illustrative example.
4.12
within the receptor’s lifetime, the risks over the lifetime are low. If the value of the K
d
parameter
is increased (i.e., its mobility decreased even further), the risks are still low because the
contaminant still has not reached the receptor. However, if the K
d
parameter is sufficiently small,
the contaminants arrive very quickly, and the receptors are exposed within their lifetime.
Different methods were presented in the previous section for determining K
d
, and 4 different
retardation factors were presented, which were based on K
d
. Because different models use
different formulations for the retardation factor, pore-water velocity, or contaminant velocity, the
same K
d
will produce different simulation results by the different models. As one can imagine,
there are no accurate generalizations that can be made with regard to defining K
d
; as such, it tends
to represent a calibration parameter in computer models. Because any modeling exercise
(conceptually, physically, and mathematically) represents a simplification of the real world, many
phenomena unknown or misunderstood are lumped into the parameters upon which modeling is
based. K
d
represents one of those parameters, where known and many unknown phenomena are
lumped; as such, K
d
tends to lose some of its meaning in the modeling world, although it retains
its full meaning in the laboratory.
In the laboratory, K
d
is determined under carefully controlled conditions; all aspects of the
experiments are controlled to ensure accuracy in the experimental exercise. The real world does
not afford the luxury of controlling the environment; therefore, the conditions surrounding the
sorption phenomenon must be estimated. Unfortunately, site conditions may not be adequately
described by sampling. Moreover, the identification of a single representative K
d
value for a site
may be impossible due to the large heterogeneities of the site’s subsurface system.
4.4.2 Partition Coefficient as a Calibration Parameter in Transport Modeling
Calibrations do not necessarily ensure that the simulated results capture the essence of the trans-
port phenomena. For example, Figures 4.2 and 4.3 present the results of a calibration using the
MEPAS model to monitored data.
1
Each figure presents time-varying
90
Sr concentrations.
Figure 4.2 presents MEPAS simulation with a K
d
equaling 0.4 ml/g (i.e., solid line and no
symbols) and a simulation with K
d
equaling 0.8 ml/g (i.e., solid line with open triangles). The
solid triangle represents the monitored data, identifying in a concentration of 220 pCi/l at year 27.
As Figure 4.2 shows, the “0.8” calibration is precise and passes directly through the monitored
data (i.e., an exact match). The “0.4” simulation does not appear to capture the essence of the
problem, as it predicts a concentration of 55,600 pCi/l at year 27. The “0.4” simulation seems to
have over predicted the concentration by over 2 orders of magnitude. The significant difference
in the peak concentration between the 2 simulations were the result of a minor change in K
d
(i.e.,
changes in the tenths place).

4.13
Figure 4.2. Example illustrating a MEPAS
90
Sr calibration with
K
d
equaling 0.8 ml/g and 1 monitored-data point.
Figure 4.3 presents the entire curve that is defined by monitored data, which includes the single
data point from Figure 4.2. The monitored data in Figure 4.3 are presented as solid circular
symbols. When all of the time-varying monitored data are considered in the assessment, the
MEPAS simulation with a K
d
equaling 0.4 ml/g appears to have captured the essence of the shape
of the monitored data more accurately than the results associated with the 0.8 ml/g simulation.
Although the “0.4” simulation does not precisely capture the entire shape of the monitored-data
curve, it has captured the peak information, which usually is considered to be most important.
Figure 4.3 illustrates the difficulty of calibrating a groundwater model to a single data point or a
set of points that are not well distributed in time. Although the “0.8" simulation captured the
single data point, it completely missed the peak concentrations and the time to peak. Because
only 1 monitored data point is used in the calibration, an unlimited number of curves could have
been simulated to pass through the monitored data point.

4.14
Figure 4.3. Example illustrating MEPAS
90
Sr calibrations with K
d
equaling 0.4 and 0.8 ml/g and several monitored-data points.
Figure 4.3 illustrates that minor changes in K
d
(i.e., in the tenths place) can result in significant
changes in simulation results. The concentrations between simulations at year 27 varied by over
2 orders of magnitude. The peak concentration decreased from 104,000 pCi/l to 31,000 pCi/l by
increasing the K
d
by only 0.4 ml/g. One simulation is over 5 times the drinking water limit of
20,000 pCi/l, while the other simulation is nearly equal to this limit. If the “0.8” results were
assumed to be correct, the assessment would have underestimated the impacts by a factor of 3.4.
Although the difference between 0.4 and 0.8 mg/l does not appear to be large, these results do
illustrate the difficulties in using K
d
and other parameters in the calibration process. These results
also suggest that the discrepancy between “calibrated” simulations and limited data could be much
larger (i.e., orders of magnitude).
4.5 Summary
Various sections in this report have illustrated that there are many definitions for K
d
(e.g., theoretical, analytical, experimental, thermodynamic, etc.), all resulting with different values.
The results presented in Figures 4.2 and 4.3 should represent a sobering reminder of the
difficulties associated with groundwater assessments using partition coefficients. In many
instances, a groundwater simulation is performed with no calibration at a site using K
d
values that
4.15
have been obtained from “peer-reviewed” literature. The analyst tries to match soil types and
environmental conditions with their site when “selecting” an appropriate K
d
. It must be
emphasized that these K
d
values are unrelated to the actual site and only represent the laboratory
conditions reported in the literature; they do not represent actual conditions at the site under
investigation. The peer-reviewed values only provided an idea as to the possible magnitude
associated with the K
d
value. Different geochemical conditions, some known and unknown, exist
between the actual site and those reported in the literature. The difficulty in using existing
literature numbers is that the phrase “peer-reviewed literature” tends to lend too much credibility
to these potentially inappropriate K
d
values.
5.1
5.0 Application of Chemical Reaction Codes
5.1. Background
Determination of species distributions for dissolved major and trace constituents, including
radionuclides, is necessary to understand the processes that control the chemistry of soil-water
systems. Several processes will control the thermodynamic activities of dissolved species and, to
some extent, their mobility in surface and ground waters and bioavailability to man. These
processes are described in detail in Chapter 2 and references cited therein. The processes include
the following:
C Aqueous complexation
C Oxidation/reduction
C Adsorption/desorption
C Mineral precipitation/dissolution
The distribution of aqueous species in a multi-component chemical system, such as those in soil-
water environments, can only be reliably calculated from a combination of accurate analyses of
water compositions and a competent chemical reaction model. Computerized chemical reaction
models based on thermodynamic principles may be used to calculate these processes depending on
the capabilities of the computer code and the availability of thermodynamic and/or adsorption data
for aqueous and mineral constituents of interest. Use of thermodynamic principles to calculate
geochemical equilibria in soil-water systems is well established and described in detail in many
reference books, such as Bolt and Bruggenwert (1978), Garrels and Christ (1965), Langmuir
(1997), Lindsay (1979), Morel (1983), Nordstrom and Munoz (1985), Sposito (1989, 1994),
Stumm and Morgan (1981), and others. The reader is referred to these sources for detailed
discussions and examples of specific applications relative to the thermodynamic principles and
equations that govern these calculations.
Because of the great importance of the aqueous speciation, adsorption, and solubility processes
relative to the concentrations and mobility of contaminants that may leach from waste, an
understanding of the capabilities and application of chemical reaction models is essential. This
understanding is additionally important because these models are used for both the scientific and
legal aspects of risk and performance assessment studies of waste disposal and mitigation of
environmental contamination.
The purpose of this chapter is to provide a brief conceptual overview of chemical reaction codes
and their use in addressing technical defensibility issues associated with data from K
d
studies.
Particular attention is given to the capabilities of EPA’s MINTEQA2 code, including the types of
conceptual models the code contains to quantify adsorption. Issues pertaining to the availability
of databases for these adsorption models and the status of the MINTEQA2 aqueous speciation
and solubility database for radionuclides are also discussed.

1
Mass transfer is the transfer of mass between 2 or more phases that includes an aqueous
solution, such as the mass change resulting from the precipitation of a mineral or adsorption of a
metal on a mineral surface. In contrast, mass transport is the time-dependent movement of one or
more solutes during fluid flow.
5.2
5.1.1 Definition of Chemical Reaction Modeling
Chemical reaction models/codes are referred to by several terms in the literature. The term may
include either of the adjectives “chemical” or “geochemical,” often depending on the technical
field of expertise of the author and/or anticipated audience. Additionally, the models/codes can be
referred to as reaction, equilibrium, speciation, or mass transfer
1
(and others) models/codes,
although some of these terms refer to submodel capabilities. Throughout this report, the terms
“chemical reaction models” and ”chemical reaction codes” will be used as collective terms for all
variations of these models and codes.
A chemical reaction model is defined here as the integration of mathematical expressions
describing theoretical concepts and thermodynamic relationships on which the aqueous speciation,
oxidation/reduction, precipitation/dissolution, and adsorption/desorption calculations are based.
A chemical reaction code refers to the translation of a chemical reaction model into a sequence of
statements in a particular computer language. We define a competent chemical reaction model as
a model that contains all the necessary submodels and important aqueous complexes, solids and
gases for the important elements of interest required to adequately interpret a given data set.
Most chemical reaction models are based on equilibrium conditions, and contain limited or no
kinetic equations in any of their submodels. Some processes, such as aqueous speciation and
cation or anion exchange, are closely approximated by equilibrium conditions over short time
frames of hours to days. On the other hand, kinetic factors may limit other processes, such as
some precipitation/dissolution and redox-sensitive reactions, from reaching equilibrium over
reaction periods of tens of years or more. Moreover, without information or assumptions
regarding the rate of release of the contaminant of interest from its source term, such as
contaminated soils or a decommissioning site, modeling calculations cannot provide an estimate of
the total mass (i.e., mass present in aqueous solution plus associated mineral phases) of a
contaminant released in the environment under review. At best, chemical modeling based on
equilibrium conditions may provide estimates of bounding limits for some processes depending on
the reactions being considered. Because of the limited availability of kinetic data and
incorporation of kinetic algorithms into chemical reaction codes, this is an important area for
future experimental studies and development of chemical reaction models. Readers are referred
to references on reviews of chemical reaction models cited later in this chapter for more details on
this issue.
Because thermodynamic data typically do not have the resolution to distinguish among different
isotopic forms of contaminant-containing aqueous species or solids, geochemical modeling
5.3
calculations do not provide any information on the distribution of the different contaminant
isotopes present in the aqueous, gaseous, or associated solid phases. However, in most
situations, radionuclide isotopes will react the same as natural (stable) isotopes of the element.
By assuming ideal isotopic mixing or exchange, one can estimate the distribution of any selected
isotopes among the bulk elemental distribution.
5.1.2 Reviews of Chemical Reaction Models
Numerous reviews of chemical reaction codes have been published. Some of the more extensive
reviews include those by Jenne (1981), Kincaid et al. (1984), Mercer et al. (1981), Nordstrom et
al. (1979), Nordstrom and Ball (1984), Nordstrom and Munoz (1985), Potter (1979), and others.
These reviews have been briefly described in Serne et al. (1990). The reviews discuss issues such
as:
C Basic mathematical and thermodynamic approaches that are required to formulate the
problem of solving geochemical equilibria in aqueous solutions
C Applications for which these codes have been developed and used, such as the modeling
of adsorption equilibria, complexation and solubility of trace metals, equilibria in brine
solutions and high-temperature geothermal fluids, mass transfer, fluid flow and mass
transport, and redox balance of aqueous solutions
C Selection of thermodynamic data and development of thermodynamic databases
C Limitations of chemical reaction codes, such as the testing of the equilibrium assumption,
application of these models to high-ionic strength aqueous solutions (e.g., the ion
association versus ion interaction conceptual models), the reliability of thermodynamic
databases, and the use of validation to identify inadequacies in the conceptual models
developed with chemical codes.
Table 5.1 provides a sampling of some chemical reaction codes that have been described in the
literature and mentioned in published proceedings, such as Erdal (1985), Jackson and Bourcier
(1986), Jacobs and Whatley (1985), Jenne (1979), Loeppert et al. (1995), Melchior and Bassett
(1990), and the reviews cited above. The reader is directed to these published proceedings and
reviews for the appropriate reference to the documentation of each code. Although this list of
chemical reaction models is not meant to be complete and continues to expand each year, it
demonstrates the diversity of codes that exist, and, in some cases, the evolution of some codes.

5.4
ADSORP
AION
ALCHEMI
AQ/SALT
ASAME
BALANCE
C-Salt
CHEMIST
CHEMTRN
CHESS
COMICS
DISSOL
ECES
ECHEM
EHMSYS
EQ3
EQ3NR
EQ6
EQBRAT
EQUIL
EQUILIB
EVAPOR
FASTCALC
FASTPATH
GEOCHEM
GEOCHEM-PC
GIBBS
GMIN
HALTAFALL
HARPHRQ
HITEQ
HYDRAQL
IONPAIR
KATKHE
KATKLE1
MICROQL
MINEQL
MINEQL2
MINTEQ
MINTEQA1
MINTEQA2
MIRE
MIX2
NOPAIR
PATH
PATHCALC
PATHI
PHREEQE
PHRQPITZ
REDEQL
REDEQL.EPAK
REDEQL2
RIVEQL
SEAWAT
SENECA
SENECA2
SIAS
SOILCHEM
SOLGASWATE
R
SOLMNEQ
SOLMNEQ.88
SOLVEQ
SYSTAB
THERMAL
WATCH1
WATCHEM
WATEQ
WATEQ2
WATEQ3
WATEQ4F
WATEQF
WATEQFC
WATSPEC
Table 5.1. Chemical reaction models described in the literature.
Nordstrom and Ball (1984) discuss the issue of why so many chemical reaction codes exist. They
attribute this diversity of codes to (1) the lack of availability, (2) inadequate documentation, (3)
difficulty of use of some chemical codes, and (4) the wide variety of calculational requirements
that include aqueous speciation, solubility, and/or adsorption calculations for aqueous systems
that range from simple, chemical systems associated with laboratory experiments to complex,
multi-component systems associated with natural environments. No single code can do all of the
desired calculations in a perfectly general way. Typically the more general and comprehensive a
geochemical code is, the more difficult and costly it is to use. Another factor may be that
scientists are inherently reluctant to use any computer code that they and their immediate
coworkers have not written.
5.1.3 Speciation-Solubility Versus Reaction Path Codes
Jenne (1981) divides chemical reaction codes into 2 general categories: aqueous speciation-
solubility codes and reaction path codes. All of the aqueous speciation-solubility codes may be

1
Complexation (i.e., complex formation) is any combination of dissolved cations with
molecules or anions containing free pairs of electrons. Species refers to actual form in which a
dissolved molecule or ion is present in solution. Definitions are taken from Stumm and Morgan
(1981).
A list of acronyms, abbreviations, symbols, and notation is given in Appendix A. A list of
definitions is given in Appendix B
5.5
used to calculate aqueous speciation/complexation,
1
and the degree of saturation (i.e., saturation
index) of the speciated composition of the aqueous solution with respect to the solids in the code's
thermodynamic database. Some aqueous speciation-solubility codes also include the capabilities
to calculate mass transfer between a single initial and final state, that results from mineral
precipitation/dissolution and/or adsorption/desorption reactions. Chemical reaction codes, such
as WATEQ, REDEQL, GEOCHEM, MINEQL, MINTEQ, and their later versions, are examples
of codes of this type.
Reaction path codes include the capabilities to calculate aqueous speciation and the degree of
saturation of aqueous solutions, but also permit the simulation of mass transfer due to mineral
precipitation/dissolution or adsorption onto adsorbents as a function of reaction progress. Typical
applications include the modeling of chemical changes associated with the interaction of a mineral
assemblage and ground water (e.g., INTERA, 1983, and Delany, 1985) or the release of
radionuclides from a proposed glass waste form (e.g., Bourcier, 1990) as a function of time.
Computationally, 1 unit of reaction progress means that 1 unit of gaseous or solid reactant (e.g.,
radioactive waste source term) has reacted with an aqueous solution in contact with solid phases
with which the solution is already in equilibrium. At each step of reaction progress, the code
calculates the changes or path of mineral and gaseous solubility equilibria that are constraining the
composition of the aqueous solution, the masses of minerals precipitated and/or dissolved to
attain equilibrium, and the resulting composition of the aqueous solution. Examples of reaction
path codes include the PHREEQE, PATHCALC, and the EQ3/EQ6 series of codes.
5.1.4 Adsorption Models in Chemical Reaction Codes
Various adsorption models have been incorporated into a small number of chemical reaction
codes to calculate the mass of a dissolved component adsorbing on a user-specified mineral phase,
such as iron hydroxide that coat mineral grains in soil. The adsorption modeling capabilities in
these codes have been briefly reviewed by others (e.g., Goldberg, 1995, and Davis and Kent,
1990) and will not be duplicated here. The options vary from code to code. Adsorption models
incorporated into chemical reaction codes include non-electrostatic, empirical models as well as
the more mechanistic and data intensive, electrostatic, surface complexation models. Examples of
non-electrostatic models include the partition (or distribution) coefficient (K
d
), Langmuir
isotherm, Freundlich isotherm, and ion exchange models. The electrostatic, surface complexation
models (SCMs) incorporated into chemical reaction codes include the diffuse layer model (DLM)

1
In general terms, the activity of an ion is its effective concentration that determines its
behavior to other ions with which it might react. The activity of an ion is equal to its
concentration only in infinitely dilute solutions, and is related to its analytical concentration by an
activity coefficient, (. Activities, activity coefficients, and associated thermodynamic relationships
are discussed in detail in texts such as Glasstone (1972), Lewis and Randall (1961), Morel (1983),
Sposito (1984), and Stumm and Morgan (1981).
5.6
[or diffuse double layer model (DDLM)], constant capacitance model (CCM), Basic Stern model,
and triple layer model (TLM).
Some of the chemical reaction codes identified in the reviews by Goldberg (1995) and Davis and
Kent (1990) as having adsorption models include HARPHRE (Brown et al., 1991), HYDRAQL
(Papelis et al., 1988), SOILCHEM (Sposito and Coves, 1988), and the MINTEQ series of
chemical reaction codes, including MINTEQA2 (Allison et al., 1991) developed for the U.S.
Environmental Protection Agency (EPA). Compared to other codes, MINTEQA2 contains some
of the most extensive options for modeling adsorption, including all of the models listed above,
except for the Basic Stern model. The MINTEQA2 adsorption model options are discussed
further in Section 5.2, and their associated equation and reaction formulations as coded within
MINTEQA2 are described in Section 5.3. It should be noted that the partition coefficient (K
d
),
Langmuir, and Freundlich models incorporated into MINTEQA2 are formulated in terms of
species activities,
1
and not the more traditional approach of total concentrations of dissolved
metal. This variation in modeling approach and the rationale for its use are discussed in
Section 5.2.
Some of these models are briefly described in Chapter 2. The reader is also referred to reference
texts by Langmuir (1997), Morel (1983), Sposito (1984), and Stumm and Morgan (1981) for
more detailed background descriptions, associated equations and data needs, and model
comparisons pertaining to these adsorption models.
As noted in Chapter 2, the electrostatic, surface complexation models, although robust, are not
expected to have a significant impact on contaminant transport and risk assessment modeling due
to their significant data needs and more complex equation formulations. Detailed descriptions,
comparisons, and derivations of the relevant equations and reactions associated with these models
are described in Westall and Hohl (1980), Morel et al. (1981), Barrow and Bowden (1987), Davis
and Kent (1990), and others. The data needs and associated derivation (i.e., parameterization) of
model constants are discussed by Morel et al. (1981), Turner (1991), and Goldberg (1995). The
electrostatic models were developed to provide a mechanistic description of adsorption reactions
in systems containing a pure single phase of an amorphous or crystalline metal oxide. Numerous
studies have demonstrated their success in predicting adsorption of trace metals in such simplified
systems (e.g., Turner, 1993). Application of such adsorption models to natural systems where the
reactive surfaces include a combination of impure phases, clays, and humic materials are limited.
The adsorption behavior of such systems unfortunately cannot be modeled assuming that the

1
The “near field” is that portion of a contaminant plume that is near the point source and whose
chemical composition is significantly different from that of the uncontaminated portion of the
aquifer. The “far field” refers to that which is not the “near field.”
5.7
adsorptive properties of a phase mixture, such as soil, can be readily predicted by adding the
adsorption constants for the individual solid phases in some normalized fashion.
Numerous papers have been published relative to the application of non-electrostatic and
electrostatic adsorption models to modeling the migration of radionuclides released from high
(HLW) and low level (LLW) radioactive waste disposal facilities. These include reviews and
references cited therein by Serne and Muller (1987) and Turner (1993,1995) for application to
HLW disposal and Serne et al. (1990) for application to LLW disposal issues. The reader should
also be aware of an extensive literature review by Berry (1992a,b,c) of adsorption studies
conducted in the United Kingdom and the international community on sorption relative to the
release and transport of radionuclides in the near
1
and far field. The literature review is published
as 3 reports. The first report summarizes studies funded by the United Kingdom (UK) Nirex and
Department of the Environment (UK DoE). The second report contains an extensive
bibliography, including reference citations and complete abstracts, of United Kingdom and
international publications on the subject area. The third report compares the objectives and
approaches used in studies funded by Nirex and UK DoE to those in related studies undertaken by
the international community.
5.1.5 Output from Chemical Reaction Modeling
The results from chemical reaction codes vary depending on the capabilities, design of the output
report, and user-selected options for each code. The output may be in the form of a report
directed to a printer, and/or a total or partial report stored as an ASCII (American Standard Code
for Information Interchange)-formatted file for future use in word processing or spreadsheet
software or as input for other scientific application software. The output can be extensive
depending on the options used for the modeling calculations and the level of output report
requested by the user.
The output report from MINTEQA2 chemical reaction code (Allison et al., 1991) will be used as
a typical example. The MINTEQA2 code was developed by EPA and is described in greater
detail in Section 5.2. For each modeling calculation, the output can include the following:
C Documentation and constraints applied to the calculation
- Name of the data file and the date and time of modeling calculations
- Documentation to describe modeling calculation
- Listing of the model parameters used to control the calculations (e.g., maximum
number of permitted iterations, method for calculating activity coefficients, alkalinity
option, units used for input of water composition, temperature), level of output report
(e.g., short versus long report), and type of selected adsorption algorithm
5.8
- Listing of the input water composition
- Listing of any controls (e.g., pH, Eh, redox equilibria) applied to the calculation
- Listing of any additions or modifications made as part of the input file to the code’s
thermodynamic database
- Listing of any adsorption reactions and associated constants used for adsorption
reaction calculations
- Listing of any solid phases and associated masses considered for mass transfer
calculations
- Listing of any gases whose solubility will control the concentration of a dissolved
constituent (e.g., solubility of CO
2
gas to fix the total concentration of dissolved
carbonate)
C Results of aqueous speciation calculations
- Number of iterations required for the aqueous speciation calculation to converge
- Calculated concentrations, activities, activity coefficients, equilibrium constants as
modified for ionic strength and temperature for each aqueous species extracted from
the code’s thermodynamic database and included in the calculation
- Charge imbalance before and after calculation of aqueous speciation
- Listing of the distribution of important (i.e., greater than 1 percent of the total
concentration of a dissolved component) uncomplexed and complexed aqueous species
for each valence form of each dissolved component (See “Glossary” for technical
definition of “component.”)
C Results of solubility calculations
- Degree of saturation of the starting water composition relative to equilibrium solubility
of every solid in the code’s thermodynamic database containing the components
included in that water analysis
- Listing of the reaction stoichiometries and associated temperature-corrected
equilibrium constants for each solid phase included in the calculation
C Results of mass transfer calculations at each stage of calculations where a solubility and/or
adsorption equilibrium condition is reached
- Repeat of all speciation results for new calculated water composition
- Repeat of the solubility results for new calculated water composition
- Calculated mass of each element in dissolved, precipitated, and/or adsorbed states for
new calculated water composition
Parts of example output reports from MINTEQ are listed and explained in detail in Allison et al.
(1991) and Peterson et al. (1987a).

1
When using the partition coefficient (K
d
) or Freundlich adsorption models, the predicted
solution-concentration limits are only valid when modeling trace concentrations of a contaminant
of interest.
5.9
5.1.6 Assumptions and Data Needs
Chemical reaction models may be used to predict the concentrations of elements, such as uranium,
that may be present in an aqueous solution. This type of modeling calculation requires the user to
select either a solubility or an adsorption reaction to constrain the maximum concentration limit of
a contaminant or any other dissolved constituent. The modeling process is based on the following
assumptions and data needs for the environment of interest:
C For a solution-concentration limit based on a solubility reaction, the mineral phase selected
as the solubility control for the contaminant of interest must have known thermodynamic
data (e.g., solubility constant). The selection of the solid phase must be technically
defensible in that the phase is known to exist in analogous aqueous environments and have
rates of precipitation and dissolution that are not limited by kinetics.
C For a solution-concentration limit based on an adsorption reaction,
1
the substrate (e.g.,
an iron-oxyhydroxide coating) selected as the adsorption control for the contaminant of
interest must be technically defensible relative to the soil or sediment being modeled. The
adsorption parameters must be known for the contaminant of interest and its major
competing ions for the substrate and the range of appropriate environmental conditions.
C The reactions or conditions that control the pH, redox conditions, and concentrations of
complexing ligands (e.g., dissolved carbonate) for the derived aqueous solution must be
assumed and technically defensible.
C The model must have an adequate thermodynamic database that includes all the necessary
aqueous species, redox reactions, minerals, and sorption substrates for the contaminant of
interest and for the other constituents of environmental importance.
C The composition of water (in particular, pH, Eh, and alkalinity) contacting the
contaminant-containing phases must be known.
C Most chemical modeling calculations will be limited to equilibrium conditions, because of
the general absence of kinetic rate values for the aqueous speciation, solubility, and/or
sorption reactions involving the contaminant of interest and other constituents of
environmental importance. Equilibrium (actually steady state) conditions are likely in the
far field, but are less likely in the near-field environment or at the boundaries of
contaminant plumes.

1
Model validation is the integrated test of the accuracy with which a geochemical model and
its thermodynamic database simulate actual chemical processes. In contrast, code verification is
the test of the accuracy with which the subroutines of the computer code perform the numerical
calculations.
5.10
5.1.7 Symposiums on Chemical Reaction Modeling
Both the diversity and interdependency of research efforts associated with chemical reaction
modeling are effectively demonstrated by the papers presented at several symposiums held on this
subject. Some of these conferences are listed in Table 5.2.
The symposiums typically include papers on a range of subjects, such as theoretical
advancements; model and code development, including documentation; application studies of
equilibrium and mass transfer codes, transport and coupled codes, and surface processes; database
development, including thermodynamic data, kinetic data, and data on organic compounds;
modeling sensitivities; and model validation.
1
The reader is encouraged to peruse these proceedings. The proceedings’ papers show that the
development of chemical reaction models is concurrent with the expansion and improvement of
thermodynamic databases for aqueous species and solids and for adsorption, as well as with
application studies that test the validity of these models and their associated databases.
Table 5.2. Examples of technical symposiums held on development,
applications, and data needs for chemical reaction modeling.
Published
Proceedings
Date of
Symposium
Location Sponsorship
Jenne (1979) Sept. 11-13, 1978 Miami Beach,
Florida
Amer. Chem. Soc.
Erdal (1985) June 20-22, 1984 Los Alamos,
New Mexico
U.S. Department of Energy (DOE) and
U.S. Nuclear Regulatory Commission (
NRC)
Jacobs and Whatley (1985) Oct. 2-5, 1984 Oak Ridge,
Tennessee
NRC
Jackson and Bourcier (1986) Sept. 14-17, 1986 Fallen Leaf Lake,
California
DOE and LLNL
Melchior and Bassett (1990) Sept. 25-30, 1988 Los Angeles,
California
Amer. Chem. Soc.
Loeppert et al. (1995)
Oct. 23-24, 1990 San Antonio,
Texas
Soil Sci. Soc. Amer. and
Amer. Soc. Agron.

1
Versions of MINTEQ modified to operate on DOS and Macintosh personal computer systems
are also available from commercial sources.
5.11
5.2 MINTEQA2 Chemical Reaction Code
5.2.1 Background
The MINTEQA2 computer code and its predecessor versions are described by Allison et al.
(1991, MINTEQA2), Brown and Allison (1987, MINTEQA1), Peterson et al. (1987a,
MINTEQ), and Felmy et al. (1984, MINTEQ). The MINTEQ code was developed with EPA
funding. It was originally constructed by combining the mathematical structure of the MINEQL
code (Westall et al., 1976) with the thermodynamic database and geochemical attributes of the
WATEQ3 code (Ball et al., 1981a).
The MINTEQA2 code is used in conjunction with a thermodynamic database to calculate
complex chemical equilibria among aqueous species, gases, and solids, and between dissolved and
adsorbed states. Conceptually, the code can be considered as having the following 4 submodels:
(1) aqueous speciation, (2) solubility, (3) precipitation/dissolution, and (4) adsorption. These
submodels include calculations of aqueous speciation/complexation, oxidation-reduction, gas-
phase equilibria, solubility and saturation state (i.e., saturation index), precipitation/dissolution of
solid phases, and adsorption. The MINTEQA2 code incorporates a Newton-Raphson iteration
scheme to solve the set of mass-action and mass-balance expressions.
The reader is referred to the references and user guides listed above for details regarding the use
of the MINTEQ code, types and examples of geochemical equilibria calculations possible with
this code, the basic equations on which the model is based, and examples of input and output files.
5.2.2 Code Availability
MINTEQA2 (Version 3.11) is the most current version of MINTEQ available from EPA. It is
compiled to execute on a personal computer (PC) using the MS-DOS computer operating system.
The MINTEQA2 software package distributed by EPA also includes PRODEFA2, which is an
user-interactive code used to create and modify input files for MINTEQA2.
1
The user is referred
to the description of PRODEFA2 in Allison et al. (1991).
Copies of the files containing the source and executable codes for MINTEQA2 and PRODEFA2,
thermodynamic databases, example input data sets, and documentation are available by mail from

1
The use of commercial business and product names is for descriptive purposes only, and does
not imply endorsement by EPA or PNNL.
Allison Geoscience Consultants, Inc., 3920 Perry Lane, Flowery Branch, Georgia 30542.
2
Environmental Education Enterprises, Inc. (E
3
), 2764 Sawbury Boulevard, Columbus, Ohio
43235-4580.
5.12
Center for Exposure Assessment Modeling (CEAM)
U.S. Environmental Protection Agency
Office of Research and Development
Environmental Research Laboratory
960 College Station Road
Athens, Georgia 30605-2720
These files may also be downloaded using the Internet by accessing CEAM’s home page. The
address of the CEAM home page is
ftp://ftp.epa.gov/epa_ceam/wwwhtml/ceamhome.htm
The MINTEQA2 code and documentation are located under “software products” in “...CEAM
software products and related descriptive information is...” The CEAM home page may also be
accessed via EPA’s home page at
http://www.epa.gov
by selecting “software” in “EPA Data Systems and Software,” and then “Center for Exposure
Assessment Modeling.”
Training courses are commonly held on the use of chemical reaction modeling techniques and the
application of the MINTEQA2 code. In the past, MINTEQ training has been provided to EPA
and NRC by their supporting national laboratory and private contractors. Allison Geoscience
Consultants, Inc.
1
have, for example, conducted several MINTEQA2 modeling workshops. The
Pacific Northwest National Laboratory (Peterson et al., 1987a) has provided MINTEQA2
training to the NRC. Short course announcements from the Environmental Education
Enterprises, Inc. (E
3
)
2
for environmental science and engineering training also included MINTEQ
workshops.
5.2.3 Aqueous Speciation Submodel
The MINTEQA2 code can be considered as having the following 4 parts: (1) an aqueous
speciation submodel, (2) solubility submodel, (3) precipitation/dissolution submodel, and

5.13
Cation/Anion Balance(%) '
[Anions (equiv./l)& Cations (equiv./l)]
[Anions (equiv./l)% Cations (equiv./l)]
×100.
(5.1)
(4) adsorption submodel. The aqueous speciation submodel is fundamental to all other
submodels. It first uses the MINTEQA2 thermodynamic database to calculate the activities of the
uncomplexed and complexed aqueous species for an initial water composition. The activities of
individual aqueous species are corrected for ionic strength using the Davies or extended Debye-
Hückel equations.
The aqueous speciation of a dissolved contaminant can only be determined using thermodynamic
calculations such as those formulated in the aqueous speciation submodel of chemical reaction
codes. Except for pH, which is the negative of the logarithm of the activity of the uncomplexed
H
+
aqueous ion, the user typically supplies the total concentrations of a chemical constituent in an
input file for a chemical reaction code. Most common analytical techniques measure the total
concentrations of a dissolved constituent such as uranium, and not the concentration of any of its
many individual species such as UO
2
2+
, UO
2
OH
+
, UO
2
(CO
3
)
2
2-
, UO
2
SO
4
"
(aq), or UO
2
PO
4
-
.
Aqueous speciation, and hence the testing of solubility hypotheses in the solubility submodel, is
only reliable if the quality of the chemical analysis of the water is adequate. The description of the
water composition is usually obtained by direct measurement of major cations and anions, pH, Eh,
and trace constituents. As a quality check of the water chemical analysis, the MINTEQA2 code
calculates the cation/anion balance for each speciated water composition. The cation/anion
balance is calculated using the equation
For simple groundwater compositions and accurate analytical work, the cation/anion balance
should not exceed a few percent (Hem, 1985).
The importance of complexation is discussed in Chapter 2 and elsewhere, such as Langmuir
(1997), Lindsay (1979), Morel (1983), and Stumm and Morgan (1981). Complexation of
dissolved metals with ligands, such as carbonate, will increase the total concentration of a
dissolved metal in a soil-water system, and affect its availability for sorption and migration in
geochemical systems. The output from the MINTEQA2 aqueous speciation submodel identifies,
based on the data in the code’s thermodynamic database, the distribution (i.e., dissolved masses)
of uncomplexed and complexed aqueous species for the constituents included in the input water
composition.
5.2.3.1 Example of Modeling Study
Krupka and Serne (1998) used the MINTEQA2 code to analyze solubility limits for contaminants
that may be released from a hypothetical low-level radioactive waste (LLW) disposal facility being
considered in a NRC performance assessment test case analysis. The species distributions plotted
for dissolved U(VI) in Figure 5.1 were taken from the MINTEQA2 calculations by Krupka and
Serne. They provide a good example of the type of information

5.14
a
4% UO
2
(CO
3
)
2
2-
91% (UO
2
)
2
CO
2
(OH)
3
-
2% UO
2
(OH)
2
o
(aq)
2% UO
2
CO
3
o
(aq)
b
91% UO
2
(OH)
3
-
9% UO
2
(OH)
4
2-
Figure 5.1. Distribution of dominant U(VI) aqueous species for
leachates buffered at pH 7.0 by local ground water
(Figure 5.1a) and at pH 12.5 by cement pore fluids
(Figure 5.1b). [Adapted from MINTEQA2 modeling
results of Krupka and Serne (1998).]
5.15
provided by the aqueous speciation submodel. Figure 5.1a shows the distribution of dominant
species (i.e., greater than 1 percent of total dissolved mass) of dissolved U(VI), respectively, for
leachates buffered at pH 7.0 by the local ground water. This distribution can be contrasted to
that in Figure 5.1b which shows the distribution of dominant U(VI) species at pH 12.5 by pore
fluids derived from ground-water interactions with cementitious materials in the hypothetical
LLW disposal facility. At pH 7.0, the speciation of dissolved U(VI) is dominated by uranyl
carbonate complexes. At very basic pH conditions, the anionic uranyl hydrolysis species dominate
the chemistry of dissolved U(VI). The speciation results clearly demonstrate that major
differences can occur in the speciation of a dissolved metal as a function of different solution
chemistries, such as pH.
5.2.3.2 Application to Evaluation of K
d
Values
As noted in Chapter 2 and published references, such as Morel (1983), Sposito (1989, 1994),
Stumm and Morgan (1981), and others, the ionic nature and composition of the dominant
aqueous species for a contaminant are important factors relative to its adsorption behavior on
reactive mineral surfaces. Moreover, as demonstrated in the example given above, the ionic
nature and composition of the dominant aqueous species are dependent on the composition, pH,
and redox conditions of a surface or ground water.
If thermodynamic data exist for the important aqueous species of a contaminant of interest,
chemical reaction models provide the most cost and time effective means of predicting the
dominant aqueous species that could exist for practically any water composition. The rate at
which these calculations can be done is limited only by the rate at which a user can enter the input
data, given the fast speeds of processors used in modern personal computers. The user can
rapidly evaluate whether the dominant species is(are) cationic or anionic, as well as how their
compositions might be affected by complexation with dissolved ligands such as carbonate and
phosphate. If there is uncertainty relative to the pH evolution or ligand content of a water, the
user may then quickly modify the input value(s) and complete a series of sensitivity analyses to
determine how the ionic charges and compositions of the dominant aqueous species change.
This information can then used to substantiate the conceptual model that is being used for
adsorption for a particular contaminant. For example, if 90 percent of the mass of a dissolved
contaminant is present in anionic form, is this consistent with low or high K
d
values that one might
find reported in the literature? If the calculations indicate strong complexation with dissolved
sulfate, are the default K
d
values in transport or risk assessment models, such as MEPAS,
conservative estimates relative to this specific site chemistry? If toxicology studies indicate that
an uncomplexed species, such as Cu
2+
, is the important actor relative to bioavailability, how does
this affect the predicted risk when the aqueous speciation calculations indicate that 99 percent of
the mass of dissolved copper is present as a carbonate complex in a given water? Chemical
reaction models provide an effective tool for calculating the responses in aqueous speciation to
different conceptual models that one might consider for soil-water systems.

1
Component (or basis) species are the “basis entities or building blocks from which all species
in the system can be built” (Allison et al., 1991). Examples include Mg
2+
, UO
2
2+
, CO
3
2-
, and SO
4
2-
for magnesium, hexavalent uranium [U(VI)], inorganic carbon, and oxidized sulfur [S(VI)],
respectively. The set of components in MINTEQA2 is predefined. They are a set of linearly
independent aqueous species in terms of which all aqueous speciation, redox, mineral, and
gaseous solubility reactions in the MINTEQA2 thermodynamic database are written.
2
Mineral solubility reactions in the MINTEQA2 database are written as formation (i.e.,
precipitation) reactions. The solubility product, K
sp,T
, (see Chapter 2), which is a commonly used
term in the literature, refers to the equilibrium constant, K
r,T
, for a mineral solubility reaction
written as a dissolution reaction.
5.16
Equilibrium: Log (IAP/K
r,T
) • 0 ,
(5.2)
Oversaturated: Log (IAP/K
r,T
) > 0 ,
(5.3)
Undersaturated: Log (IAP/K
r,T
) < 0 ,
(5.4)
For example, Kaplan et al.(1998) conducted laboratory batch K
d
experiments to study the effects
of background geochemistry on the sorption of U(VI) on natural sediments. The MINTEQA2
code was used to calculate the aqueous speciation of U(VI) in a groundwater before and after
equilibrium with sediments. The modeling results indicated dissolved U(VI) was present as
essentially all aqueous anionic U(VI)-carbonate complexes [e.g., UO
2
(CO
3
)
3
4-
] at high pH
conditions. Studies by Waite et al. (1994) and others have shown that these complexes, due to
their anionic nature, tend to sorb appreciably less to sediments than cationic U(VI) complexes
which are present at lower pH conditions.
5.2.4 Solubility Submodel
After calculating the aqueous speciation for a given water composition, solubility-equilibria
hypotheses are tested. Ion activity products (IAP) are calculated from the activities of the
component (or basis) species,
1
using the stoichiometries of the solubility reactions for minerals
and other solids in the thermodynamic database. These activity products are then compared to
the equilibrium constants (K
r,T
)
2
stored in the database for the solubilities for the same solids, to
test the assumption that certain of the dissolved constituents in the aqueous solution are in
equilibrium with particular solid phases. Saturation indices, [log (IAP/K
r,T
)], are calculated to
determine if the water is at
or
with respect to a specified solid phase. This information allows one to ascertain permissible
equilibrium solubility controls for dissolved constituents in that water. This water may be a
surface or ground water, or a laboratory solution used for solubility or K
d
measurements.

1
Although Krupka et al. (1983) used the WATEQ4 chemical reaction code, their results are
analogous to the types of saturation index calculations permitted with the MINTEQ2A code.
5.17
-2.0
-1.0
0.0
1.0
2.0
2.0 3.0 4.0 5.0 6.0 7.0
pH
Saturation Index, Log IAP/K
5.2.4.1 Example of Modeling Study
Figure 5.2 shows the saturation indices calculated by Krupka et al. (1983)
1
for the mineral
rutherfordine (UO
2
CO
3
) for published analyses of solution samples taken from laboratory
uranium solubility studies. The saturation index results demonstrate that these solution samples
calculate to be at or very near equilibrium with respect to rutherfordine based on the available
thermodynamic data for this mineral and U(VI) aqueous species included in the modeling
calculations. Rutherfordine may have therefore precipitated during the course of the solubility
studies reported in the cited literature.
Figure 5.2. Saturation Indices calculated for rutherfordine (UO
2
CO
3
)
as a function of pH for solution analyses from
Sergeyeva et al. (1972). [Adapted from WATEQ4
modeling results of Krupka et al. (1983). The filled
square and triangle symbols refer, respectively, to
solutions analyses from 25 and 50
"
C experiments by
Sergeyeva et al. (1972)]
5.18
5.2.4.2 Application to Evaluation of K
d
Values
Chemical reaction codes can be used to analyze the adequacy of laboratory measurements of K
d
values for a particular soil-water system. As noted in Chapter 3, solubility limits have sometimes
been exceeded during the process of making laboratory measurements of K
d
values. This can
result when the concentration of the contaminant spike introduced to the equilibration vessel is
too great and/or when the initial chemical conditions, such as pH, vary greatly during the course
of the measurements.
By modeling the aqueous speciation and saturation indices for the initial and final compositions of
aqueous solutions present in the K
d
experiments, the user can test if any solubility limits were
exceeded during the measurements. In those cases where a contaminant-containing solid is
precipitated, the determined K
d
values are measurements of both solubility and adsorption
processes and will result in an over-prediction of contaminant attenuation (via only adsorption
processes) in the soil-water system.
Kaplan et al. (1998) conducted laboratory batch K
d
experiments to study the effects of
background geochemistry on the sorption of U(VI) on natural sediments. MINTEQA2
calculations indicated that dissolved U(VI) was present as essentially all aqueous anionic U(VI)-
carbonate complexes. Waite et al. (1994) and others have shown that these complexes, due to
their anionic nature, tend to sorb appreciably less to sediments than cationic U(VI) complexes
present at lower pH values. However, the K
d
values measured by Kaplan et al. (1998) increased
from 1.07 to 2.22 ml/g as the pH increased from 8.17 to 9.31, and were >400 ml/g at pH$10.3.
Kaplan et al. (1998) used MINTEQA2 saturation index calculations to show that the apparent
increase in U(VI) K
d
values was due to the precipitation of uranium-containing solids and not to
U(VI) adsorption to the sediment.
5.2.5 Precipitation/Dissolution Submodel
The results from the solubility model are in turn used by the MINTEQA2 as input for the
precipitation/dissolution submodel. Application of this submodel is optional. The user may select
this submodel and its different options to predict the mass of a solid phase(s) that precipitates or
dissolves in the modeled system. The mass transfer submodel determines the mass of a solid
phase(s) (e.g., a contaminant-containing solid or a mineral present in a soil) that precipitates from
a ground water or dissolves from a soil-water system. If a given water composition calculates to
be oversaturated, [log (IAP/K
r,T
) > 0], with respect to a solid phase(s) considered in the modeling
problem, the mass transfer model will decrease (i.e., precipitate a solid phase) the masses of the
appropriate dissolved constituents until the water composition is at equilibrium,
[log (IAP/K
r,T
) = 0], with respect to that solid phase(s). The MINTEQA2 output lists the mass of
solid precipitated per a set volume of the system being modeled. If a given water composition
calculates to be undersaturated, [log (IAP/K
r,T
) < 0], with respect to a solid phase(s) selected in
the modeling problem, the mass transfer model will increase (i.e., dissolve a solid phase) the
5.19
masses of the appropriate dissolved constituents until the water composition is at equilibrium with
respect to that solid phase(s) or until the user-specified finite mass of that solid has been
completely dissolved. For those solids originally designated as having finite masses, the
MINTEQA2 output gives the masses per set system volume of any of these solids remaining at
final equilibrium.
5.2.5.1 Example of Modeling Study
The solubility limits calculated by Krupka and Serne (1998) demonstrate one of several
applications for a precipitation/dissolution submodel. Maximum concentration limits for dissolved
americium, neptunium, nickel, plutonium, radium, strontium, thorium, and uranium were
calculated using MINTEQA2 for 2 ground-water environments associated with a hypothetical
LLW disposal system. The 2 limiting environments included: (1) a cement buffered system,
wherein the leachate pH is controlled at values above 10 by the effective buffering capacity of the
concrete, and (2) a ground-water buffered system, wherein the leachate pH and related solution
parameters are dominated by the local ground-water system.
Figure 5.3 shows the maximum concentrations calculated by Krupka and Serne (1998) for total
dissolved uranium as a function of pH. The predicted concentration limits are based on the
equilibrium solubilities of schoepite [UO
2
(OH)
2
·H
2
O] and uranophane
[Ca(H
3
O)
2
(UO
2
)
2
(SiO
4
)
2
·3H
2
O]. These 2 solids were selected based on published phase-stability
information and knowledge of the geochemistry of contaminant aqueous systems. Schoepite
precipitates readily in short-duration laboratory experiments conducted at ambient temperatures.
Because the concentration of dissolved uranium in equilibrium with schoepite is higher than the
solubilities of other uranium solids that precipitate under these conditions or in nature,
concentration limits based on schoepite are therefore expected to be highly conservative. The
presence of alkali and/or alkaline earth ions at high pH conditions results in the precipitation of
alkali/alkaline earth uranyl compounds that control the solubility of uranium at concentrations
lower than those resulting from equilibrium with schoepite. Therefore, the solubility of
uranophane may provide a more realistic solubility limit for dissolved uranium, especially at high
pH conditions. Uranophane is known to exist in uranium-loaded cementitious mixtures and thus
may be a realistic solubility control for dissolved uranium in cement dominated systems.
5.20
-10.0
-8.0
-6.0
-4.0
-2.0
0.0
5 6 7 8 9 10 11 12
pH
Log Uranium Concentration (mol/l)
Schoepite
Uranophane
Figure 5.3. Maximum concentration limits calculated for total dissolved
uranium as a function of pH based on the equilibrium
solubilities of schoepite and uranophane.
5.2.5.2 Application to Evaluation of K
d
Values
Chemical reaction codes can be used to calculate bounding, technically-defensible maximum
concentration limits for dissolved contaminants as a function of key composition parameters
(e.g., pH) of any specified soil-water system. The concentration of a dissolved contaminant
predicted with default or site specific K
d
values used in transport or risk assessment models may
exceed the concentration limit based on solubility relationships. In these instances, the solubility-
limited concentration may provide a more realistic bounding value than one based on a K
d
value
for the assessment calculation, and could have an important impact on the estimated level of risk.
If a calculated concentration limit is based on the solubility of a mineral that is known to
precipitate under analogous chemical conditions and over reasonable time frames, then the user
knows that the dissolved concentrations of this contaminant in an actual, open soil-water system
cannot exceed these values and will most likely be significantly less than these values due to
adsorption and/or coprecipitation processes.
Moreover, as with the aqueous speciation calculations discussed in Section 5.2.3, mass transfer
calculations can be rapidly and inexpensively repeated using a chemical reaction code to determine
5.21
their sensitivity to a wide range of chemical parameters for a soil-water systems. This includes
easily measured parameters, such as pH, and analytical values that might have a wide range of
uncertainty, such as the concentration of a dissolved complexant.
5.2.6 Adsorption Submodel
The MINTEQA2 also includes a submodel to calculate the adsorption of dissolved constituents
onto the surfaces of solid phases that can be selected by the code user. The MINTEQA2 code
includes 7 adsorption model options. These are:
C Non-electrostatic adsorption models
- Activity partition coefficient (K
d
act
) model
- Activity Langmuir model
- Activity Freundlich model
- Ion exchange model
C Electrostatic adsorption models
- Constant capacitance model (CCM)
- Diffuse layer model (DLM)
- Triple layer models (TLM).
The equations and reactions that support these models, as coded in MINTEQA2, are described in
greater detail in Section 3. These descriptions and associated equations are adapted from
Allison et al. (1991).
The K
d
act
, Langmuir, and Freundlich models in MINTEQA2 are formulated in terms of species
activities, and not the more traditional approach of total concentrations of dissolved metal. In the
latter case, the total concentrations of a dissolved metal M would equal the sum of the
concentrations of all of its dissolved complexed and uncomplexed species. For example, using the
species listed in the MINTEQA2 thermodynamic database, the total concentrations of dissolved
cadmium, [Cd]
total
, in the absence of any organic complexants in the water, could include the
following species:
[Cd]
total
= [Cd
2+
] + [CdOH
+
] + [Cd(OH)
2
"
(aq)] + [Cd(OH)
3
-
] +
[Cd(OH)
4
"
(aq)] + [Cd
2
OH
3+
] + [CdCO
3
"
(aq)] + [Cd(CO
3
)
3
4-
] +
[CdHCO
3
+
] + [CdNO
3
+
] + [CdSO
4
"
(aq)] + [Cd(SO
4
)
2
2-
] +
[CdHS
+
] + [Cd(HS)
2
"
(aq)] + [Cd(HS)
3
-
] + [Cd(HS)
4
2-
] +
[CdCl
+
] + [CdCl
2
"
(aq)] + [CdCl
3
-
] + [CdOHCl
"
(aq)] +
[CdF
+
] + [CdF
2
"
(aq)] + [CdBr
+
] + [CdBr
2
"
(aq)] +
[CdI
+
] + [CdI
2
"
(aq)].
5.22
In the presence of organic complexants, [Cd]
total
could also include, in addition to the cadmium
species listed above, the concentrations of aqueous cadmium complexes containing citrate,
acetate, EDTA, HEDTA, or other organic complexes.
A total concentration approach would therefore assume that all species of metal M absorb with
equal strength. Experimental data suggest, however, that only certain aqueous species react with
the surfaces of a mineral [e.g., Waite et al. (1994)]. Based on this assumption, these non-
electrostatic models have been reformulated, as those coded in MINTEQA2, in terms of the
activities of adsorbing species to provide activity-based models. The purpose of this approach is
to reduce the dependency of the model parameters to effects from ionic strength and aqueous
complexation of the adsorbing metal by effectively allowing the adsorption of only selected
aqueous species of each metal.
Limitations remain, however, regarding these activity formulations of the K
d
act
, Langmuir, and
Freundlich models which restricts their range of applicability. These non-electrostatic adsorption
models do not consider: charge balance on surface sites and adsorbed species, electrostatic forces
between the adsorbing species and charge surface of the mineral, and reactions between the
mineral and dissolved constituents other than the adsorbing metal. The effect of these processes
changes with variations in the composition of an aqueous solution. These processes are,
however, incorporated into the more robust, but more data intensive, electrostatic “surface
complexation” adsorption model options in MINTEQA2.
The MINTEQA2 code includes the reaction components and formalisms necessary to enter the
required adsorption data for any of the adsorption models. The code does not however have an
adsorption database for these models. The user must provide the set of surface reactions and the
associated equilibrium constants as part of the input data set. MINTEQA2 requires that this
information be supplied relative to the adsorption of constituents onto specific mineral phases,
such as amorphous ferric hydroxide [Fe(OH)
3
(am)], and not a multi-mineral phase material, such
as a soil or crushed rock. Examples of MINTEQA2 input files that include the adsorption
modeling option are included in the data files distributed by EPA, and are also listed in Allison et
al. (1991 Appendix D) and Peterson et al. (1987a). These examples demonstrate the major data
requirements for some of the adsorption model options in MINTEQA2.
5.2.6.1 Examples of Modeling Studies
Modeling studies by Peterson et al. (1986), Davis and Runnells (1987), Loux et al. (1989), and
Turner et al. (1993) are examples of the use of MINTEQ adsorption model options. Peterson et
al. (1986) and Davis and Runnels (1987) studied ground-water contamination associated with
waste impoundments for uranium mill tailings using laboratory and computer modeling
techniques. Peterson et al. (1986) modeled the adsorption of arsenic, chromium, lead, selenium,
and zinc using the triple layer model (TLM) in MINTEQ. Their conceptual model was based on
the assumption that adsorption of these metals occurred only on amorphous ferric hydroxide
[Fe(OH)
3
(am)] that precipitated and dissolved during the course of their experiments.
5.23
Adsorption parameters for the TLM for amorphous ferric hydroxide were taken from published
sources. The results of the adsorption calculations were in good agreement with some results
from their laboratory experiments.
Davis and Runnels (1987) used MINTEQ to successfully model the behavior of zinc observed in
laboratory column experiments. They assumed that the concentration of dissolved zinc measured
in their solution samples was controlled by adsorption on amorphous ferric hydroxide
[Fe(OH)
3
(am)] that precipitated as a result of pH changes occurring in their experiments. The
adsorption of zinc on Fe(OH
3
) (am) was calculated using the TLM in MINTEQ. Davis and
Runnels describe the selection of adsorption parameters used for the TLM.
Loux et al. (1989) used the MINTEQA2 code to model the pH-dependent partitioning
of 8 cationic constituents by precipitation and/or adsorption on a sandy aquifer material in an
oxidizing environment. The constituents of interest included barium, beryllium, cadmium, copper,
nickel, lead, thallium, and zinc. Adsorption of these elements was based on amorphous iron oxide
as the only reactive adsorption surface and calculated using the diffuse layer model (DLM). The
adsorption parameters and associated reactions for the diffuse layer model were taken from
Dzombak (1986). The modeling results were compared to laboratory data for the aquifer material
spiked with the trace metals. The predicted concentrations based on the diffuse layer model for
adsorption of lead, nickel, and zinc, provided a good description of the pH behavior observed for
the spiked samples. The concentrations of the other trace metals were not adequately predicted
by the model. These differences were attributed to limitations in the model and/or available
thermochemical data.
Turner et al. (1993) used the TLM in MINTEQA2 code to model adsorption data for U(VI) on
goethite ["-FeO(OH)]. The FITEQL code was used for adsorption parameter optimization.
Their study illustrates the extensive parameter-fitting process that the user must complete to use
complex electrostatic adsorption models, such as the TLM.
5.2.6.2 Application to Evaluation of K
d
Values
Chemical reaction models cannot be used to predict a K
d
value. The user must supply the
adsorption parameters when using any of the adsorption model options. Typically, the data
required to derive the adsorption parameters needed as input for adsorption submodels in
chemical reaction codes are more extensive than information reported in a laboratory batch K
d
study. However, if the parameters have been determined for a particular constituent for a surface
complexation model, a chemical reaction model, such as MINTEQA2, can be used to calculate
the masses of a constituent that are dissolved or adsorbed and how changes in geochemical
conditions, such as pH, affect its adsorption behavior. The user can then derive a K
d
using the
calculated dissolved and adsorbed masses of the constituent.
The EPA (EPA, 1992a, 1996) has used the MINTEQA2 model and this approach to estimate K
d
values for several metals under a variety of geochemical conditions and metal concentrations to
5.24
support several waste disposal issues. The EPA in its “Soil Screening Guidance” determined
MINTEQA2-estimated K
d
values for barium, beryllium, cadmium, Cr(III), Hg(II), nickel, silver,
and zinc as a function of pH assuming adsorption on a fixed mass of iron oxide (EPA, 1996; RTI
1994). The calculations assumed equilibrium conditions, and did not consider redox potential or
metal competition for the adsorption sites. In addition to these constraints, EPA (1996) noted
that this approach was limited by the potential sorbent surfaces that could be considered and
availability of thermodynamic data. Their calculations were limited to metal adsorption on iron
oxide, although sorption of these metals to other minerals, such as clays and carbonates, is well
known.
The data needed to use surface complexation adsorption models are more extensive than those
from K
d
studies. More importantly, the data for surface complexation models are based on
adsorption on pure mineral phases, such as "-Al
2
O
3
, (-Al
2
O
3
, böhmite, goethite, magnetite,
lepidocrocite, ferrihydrite, SiO
2
, biotite, or kaolinite. Natural soils are more complicated,
commonly containing mixtures of more than 10 pure minerals and amorphous mineral coatings.
Unless a user can technically defend the assumption that the adsorption of a specific contaminant
is dominated in a specific soil-water system, for example, by goethite reactive surfaces, the user is
still left with the challenge of extrapolating these modeling results for pure mineral substrates to
complex heterogeneous soil-water systems. This issue has been and will continue to be the
subject of intensive study, but is not likely to be resolved in the short term or impact contaminant
migration and risk assessment modeling soon.
5.2.7 MINTEQA2 Databases
The MINTEQA2 model includes an extensive thermodynamic database that is integrated with the
aqueous speciation, solubility, and precipitation/dissolution submodels. The content and
equations governing the values stored in the thermodynamic database are described below.
MINTEQA2 does not have per se an integrated adsorption submodel database. The adsorption
reactions and associated model parameters are supplied by the user as part of each input file.
However, as discussed below, the current MINTEQA2 software package is supplied with a
limited data file for the diffuse layer model (DLM).
5.2.7.1 Thermodynamic Database
The MINTEQA2 thermodynamic database is considered by many to be one of the most extensive
databases for modeling the aqueous speciation and solubility of contaminants and geologically-
significant constituents (e.g., magnesium, silica, aluminum, etc.) in low-temperature, soil-water
systems. To understand the fundamental data needs for a thermodynamic database of a chemical
reaction code, the basic equations underlying the thermodynamic parameters stored in the
MINTEQA2 thermodynamic database will be reviewed in the next section. The content of the
MINTEQA2 thermodynamic database as distributed by EPA will be reviewed and then compared
relative to the priority constituents considered in the scope of work for this project.

5.25
log K°
r,T
'
& )G°
r,T
2.303 R T
(5.5)
)G°
r,298
' 3 )G°
f ,298
(products) & 3 )G°
f,298
(reactants) .
(5.6)
log K°
r,T
' log K°
r,298
&
)H°
r,298
2.303 R
(
1
T
&
1
298
) .
(5.7)
)H°
r,298
' 3 )H°
f,298
(products) & 3 )H°
f,298
(reactants) .
(5.8)
log K°
r,T
. log K°
r,298
.
(5.9)
5.2.7.1.1 Basic Equations
Thermodynamic data used by MINTEQA2 are stored in the form of equilibrium constants (K
r
°
,298
)
and enthalpies (heats) of reaction (ªH
r
°
,298
) for aqueous speciation, oxidation/reduction, mineral
solubility, and gas solubility reactions. The reference temperature for the MINTEQA2 database,
as with most geochemical models, is 298 K (25 °C). Equilibrium constants (log K
r
°
,T
) may be
based on values that have been experimentally-determined or calculated from Gibbs free energies
of reaction (ªG
r
°
,T
) units of cal/mol according to the equation:
where T = temperature in degrees Kelvin,
R = gas constant (1.9872 cal/mol·K)
Values for ªG
r
°
,T
are calculated from published values for the Gibbs free energy of formation
(ªG
f
°
,298
) for each product and reactant in the aqueous speciation or solubility reaction by the
equation:
To calculate aqueous speciation and solubilities at temperatures other than 25°C, the equilibrium
constants are recalculated by the MINTEQA2 code to the temperature T of interest using the
van't Hoff relation:
Values for enthalpies of reaction are calculated from published enthalpy of formation values
(ªH
f
°
,298
) using the equation:
Values for ªH
r
°
,298
cannot be calculated for some reactions, because ªH
f
°
,298
values have not been
determined for 1 or more reaction products and/or reactants. In these cases, the MINTEQA2
code assumes that
Because of the limitations in using the van't Hoff relation for extrapolations over a wide range of
temperature, applications of the MINTEQA2 code are limited to temperatures less than 100°C.
5.26
5.2.7.1.2 Structure of Thermodynamic Database Files
Typically, each aqueous species, redox, mineral, and gas solubility reaction is represented by 2 fix-
formatted lines in the thermodynamic database files supplied with MINTEQA2. A third line is
sometimes included when the stoichiometry of a reaction is complex. The first file line includes
the identification number, formula descriptor, ªH
r
°
,298
(if available), log K
r
°
,298
, charge, and related
data for each reaction. The second line includes the reaction stoichiometry information
formulated in terms of the MINTEQA2 components. Each reaction is entered as a formation
reaction; that is, the components react to form the “more complex” species, such as an aqueous
complex or mineral phase. The hydrogen stoichiometric component of each reaction is balanced
with the components H
+
and H
2
O. The hydroxyl species, OH
-
, is not used as a component, but is
“formed” in a separate reaction in MINTEQA2.
Based on the protocol used for the MINTEQA2 thermodynamic database, the formation reaction
for the uranyl mixed hydroxide/carbonate aqueous species, (UO
2
)
2
CO
3
(OH)
3
-
, is
2 UO
2
2+
+ CO
3
2-
+ 3 H
2
O = 3 H
+
+ (UO
2
)
2
CO
3
(OH)
3
-
.
The corresponding entry in the MINTEQA2 database (in fixed format fields) for this reaction and
its associated thermochemical data is
8931405 UO2)2CO3OH)3 -14.3940 -0.8969 0.000 0.000-1.00 4.00 0.00 651.0868
2.00 4 2.000 893 1.000 140 3.000 2 -3.000 330
For more detailed format information on the MINTEQA2 database files, the reader is referred to
the documentation in Allison et al. (1991, Appendix A).
5.2.7.1.3 Database Components
The thermodynamic database in the original MINTEQ code (Felmy et al., 1984) was taken from
the WATEQ3 code (Ball et al., 1981a). Therefore, many of the inorganic reactions and
associated thermodynamic values in the MINTEQA2 database can be traced back to the database
supplements and sources described in publications documenting the WATEQ series of chemical
reactions codes (Ball et al., 1981a, WATEQ2; Ball et al., 1981b, WATEQ3; Plummer et al.,
1976, WATEQ; Truesdell and Jones, 1973, WATEQ; Truesdell and Jones 1974, WATEQ).
The thermodynamic database of the current version of MINTEQA2 includes the original
MINTEQ database plus modifications and additions completed on contracts with EPA funding.
Some of these supplements include, for example, those completed at PNNL, such as the addition
of reactions for aqueous species, gases, and solids containing cyanide and antimony by Sehmel

1
Deutsch, W. J., and K. M. Krupka. September 1985. MINTEQ Geochemical Code:
Compilation of Thermodynamic Database for the Aqueous Species, Gases, and Solids Containing
Chromium, Mercury, Selenium, and Thallium. Unpublished report prepared by Pacific Northwest
Laboratory for the U.S. Environmental Protection Agency in Athens, Georgia.
2
Although important to contaminant disposal and remediation activities (i.e., “mixed wastes”)
in the United States, computer modeling of the complexation of contaminant metals with organic
complexes was excluded from the scope of the current project due to funding limits.
5.27
(1989) and those containing chromium, mercury, selenium, and thallium by Deutsch and Krupka.
1
Documentation for these database supplements are not listed in Brown and Allison (1987,
MINTEQA1) or Allison et al. (1991, MINTEQA2), and may not be publicly available.
The elements for which the MINTEQA2 thermodynamic database has aqueous speciation, mineral
solubility, and/or gas solubility reactions are listed in Table 5.3. The second and third columns of
this table list the component species used for these elements and the redox reactions, if any,
included in MINTEQA2 for different valence states of a particular element. The reader should
note that the database does not contain reactions and associated thermodynamic values for
specific isotopes of a particular element. The calculated reactions for a soil-water system assumes
the total mass of each element.
Although the list of elements in Table 5.3 is substantial, this table and/or a listing of the database
files does not indicate if the database of a chemical reaction code, especially for key contaminants,
is adequate (i.e., completeness of reactions and quality of associated thermodynamic values) and
up-to-date. The user essentially has this important responsibility. One should expect that, as the
period of time between the publication of a code’s documentation and its use in an application
study increases, the thermodynamic database becomes dated and revisions may be warranted.
Table 5.4 lists the organic ligands for which the MINTEQA2 thermodynamic database has
aqueous speciation reactions.
2
Because of the limited availability of thermodynamic data for
metal-organic complexes important to contaminated soil-water systems, as compared to
inorganic aqueous complexes, the MINTEQA2 database, as with all chemical reaction codes, is
limited. It does not contain complexation reactions for all metals with each of the organic ligands
listed in Table 5.4. The reader will need to do a computer search of the MINTEQA2 ASCII file
containing these reactions to determine the extent of the organic complexation reactions for each
metal.
5.2.7.1.4 Status Relative to Project Scope
The contaminants chosen for study in this project include chromium, cadmium, cesium, tritium
(
3
H), lead, plutonium, radon, strontium, thorium, and uranium. Because the MINTEQA2
thermodynamic database does not contain reactions for specific isotopes, an appraisal of the
database content to aqueous speciation and solubility reactions containing tritium is not

1
Deutsch, W. J., and K. M. Krupka. September 1985. MINTEQ Geochemical Code:
Compilation of Thermodynamic Database for the Aqueous Species, Gases, and Solids Containing
Chromium, Mercury, Selenium, and Thallium. Unpublished report prepared by Pacific Northwest
Laboratory for the U.S. Environmental Protection Agency in Athens, Georgia.
5.28
appropriate. As will be discussed in Volume II of this report, the concentrations of dissolved
tritium will be affected by exchange reactions involving hydrogen-containing species dissolved in
the soil-water system.
Of the remaining elements, the MINTEQA2 thermodynamic database contains aqueous speciation
and solubility reactions for chromium, including the valence states Cr(II), Cr(III), and Cr(VI);
cadmium; lead; strontium; and uranium, including the valence states U(III), U(IV), U(V), and
U(VI). Except for uranium, the adequacy of the database for these listed elements is not known.
Data supplied by Deutsch and Krupka
1
in 1985 is the probable basis for the chromium reactions
and associated thermodynamic data. The reactions for cadmium, lead, and strontium may be
those taken from the WATEQ-series of codes and supplied with the original MINTEQ code by
Felmy et al. (1984). It is not known if these have been revised or supplemented since that time.
The reactions and associated thermodynamic data for uranium aqueous species and solid phases
were those supplied with the original MINTEQ code. They were taken from those added to
WATEQ3 (Ball et al., 1981a) and are based primarily on the compilation of uranium
thermodynamic data by Langmuir (1978). Langmuir’s review has been superseded by the
comprehensive review and compilation of uranium thermodynamic data given in Wanner and
Forrest (1992). This compilation represents a significant improvement and update to the values in
Langmuir et al. (1978), including for U(VI) carbonate and hydrolysis species that are important in
soil-water systems with pH values greater than 5.
Of the elements included in the project scope, the thermodynamic database distributed by EPA
with MINTEQA2 does not contain reactions and associated thermodynamic data for aqueous
species and solids containing cesium, plutonium, radon, and thorium. Published compilations of
thermodynamic data for aqueous species, solids, and gases containing these elements are
available, such as an Langmuir and Herman (1980), Lemire and Tremaine (1980), Peterson et al.
(1987b), Phillips et al. (1988), Smith and Martell (1976 and more recent supplements), Smith et
al. (1997), Wagman et al. (1982), and others. These sources can be used as good starting points
for adding reactions for cesium, plutonium, radon, and thorium to the MINTEQA2 database.
However, because these sources are becoming dated, additional reviews of the more recent
thermodynamic literature would be needed to supplement them and generate more up-to-date
compilations for these elements. Other compilations of thermodynamic data for these elements
include databases compiled by geochemical modeling groups elsewhere in the United States and
other countries. When documented, these databases are useful sources of information.

5.29
Table 5.3. Component species in MINTEQA2 thermodynamic database.
Element Component Species Valence States
Ag Ag
+
Al Al
3+
As H
3
AsO
3
E (aq), H
3
AsO
4
E, (aq) As(III), As(V)
B H
3
BO
3
E (aq)
Ba Ba
2+
Br Br
-
C CO
3
2-
, CN
-
, OCN
-
Ca Ca
2+
Cd Cd
2+
Cl Cl-
Cr Cr
2+
, Cr(OH)
2
+
, CrO
4
2-
Cr(II), Cr(III), Cr(VI)
Cu Cu
+
, Cu
2+
Cu(I), Cu(II)
F F
-
Fe Fe
2+
, Fe
3+
Fe(II), Fe(III)
Electron e
-
H H
+
, H
2
O (l)
Hg Hg
2
2+
, Hg(OH)
2
E (aq) Hg(I), Hg(II)
I I
-
K K
+
Li Li
+
Mg Mg
2+

5.30
Table 5.3. Continued.
Element Component Species Valence States
Mn Mn
2+
, Mn
3+
Mn(II), Mn(III)
N
NH
4
+
, NO
2
-
, NO
3
-
, CN
-
,
OCN
-
N(-III), N(III), N(V)
Na Na
+
Ni Ni
2+
P PO
4
3-
Pb Pb
2+
Rb Rb
2+
S HS
-
, SE, SO
4
2-
S(-II), S(VI)
Sb Sb(OH)
3
E (aq), Sb(OH)
6
-
Sb(III), Sb(V)
Se HSe
-
, HSeO
3
-
, SeO
4
2-
Se(-II), Se(IV), Se(VI)
Si H
4
SiO
4
E (aq)
Sr Sr
2+
Tl Tl
+
, Tl(OH)
3
E (aq) Tl(I), Tl(III)
U U
3+
, U
4+
, UO
2
+
, UO
2
2+
U(III), U(IV), U(V), U(VI)
V V
2+
, V
3+
, VO
2+
, VO
2
+
V(II), V(III), V(IV), V(V)
Zn Zn
2+

1
Center for Nuclear Waste Regulatory Analyses (CNWRA), Southwest Research Institute,
San Antonio, Texas
5.31
Table 5.4. Organic ligands in MINTEQA2 thermodynamic database.
Organic Constituents / Complexants
acetate
butyrate
iso-butyrate
citrate
diethylamine
dimethylamine
EDTA
4-
ethylenediamine
formate
fulvate
glutamate
glycine
hexylamine
humate
iso-propylamine
n-propylamine
methylamine
2-methyl pyridine
3-methyl pyridine
4-methyl pyridine
n-butylamine
nitrilotriacetate
3-
phthalate
propanoate
salicylate
tartrate
tri-methylamine
tributylphosphate
valerate
iso-valerate
It should be noted that the thermodynamic database distributed with EPA’s MINTEQA2 software
package does not include reactions and thermodynamic data for aqueous species and solids
containing americium, cobalt, neptunium, niobium, radium, and technetium. Although these
radionuclides are not part of the scope of this project, they may be important with respect to
contamination and remediation at some sites in the United States and/or performance assessments
of proposed LLW and HLW disposal facilities or decommissioning sites. Except for niobium,
published compilations of thermodynamic data for these elements, especially for americium (Silva
et al., 1995) and technetium (Rard, 1983), exist that can be used to supplement the MINTEQA2
database. The thermodynamic data for aqueous species and solids containing niobium are
extremely limited which precludes adequate modeling of aqueous/solid phase equilibria for
niobium in soil-water systems.
The thermodynamic database of MINTEQA2 was augmented by Krupka and Serne (1998) for
aqueous species and solids containing several radionuclide elements of interest to NRC. These
database modifications were based on data files provided by D. Turner
1
who had added these
5.32
reactions and thermodynamic data to his version of MINTEQA2. The database additions
included MINTEQ-formatted reactions, associated thermodynamic data (i.e., log K°
r,298
and
)H°
r,298
) and ancillary information (e.g., identification number, formula, charge, mass, reaction
stoichiometry) for aqueous species and solids containing americium, neptunium, plutonium,
radium, technetium, thorium, and uranium. The database changes for uranium are based on the
compilation by Wanner and Forest (1992), and supersede those listed in the MINTEQA2 database
as obtained from EPA. Additional revisions to the thermodynamic data for these radionuclide
elements were identified by Krupka and Serne (1998) and added to the MINTEQA2 files. These
database modifications have not undergone an in depth examination relative to quality-assurance
considerations.
5.2.7.1.5 Issues Related to Database Modifications
Successful application of chemical reaction models to quantify contaminant release and transport
in soil-water systems is dependent on the development of adequate and internally consistent
thermodynamic databases. The thermodynamic databases of chemical reaction codes are typically
revised or supplemented based on specific project needs and the availability of thermodynamic
data for aqueous species, gases, and solids containing the constituents of interest.
Although an extensive number of tabulations and critical reviews [e.g., see references in Serne et
al. (1990, Table 3.2)] of thermodynamic data for inorganic complexes and solids have been
published during the last 20 years, the selection of "best" values from these publications is a
technically and logistically challenging effort. Some of the issues and problems associated with
the selection of thermodynamic data are described in detail in Potter (1979), Nordstrom and
Munoz (1985), and Smith and Martell (1995). The critical evaluation and selection of a
thermodynamic database requires an understanding of general solution chemistry and the phase
assemblages of minerals and related amorphous solids associated with a particular cationic and/or
anionic constituent. The investigator developing the model's database
must also be cognizant of the criteria initially used to review and select the original data for the
published tabulations.
Because thermodynamic data tabulations usually contain an inadequate amount of reviewed
thermodynamic data for aqueous species of trace metals, available tabulations are typically
deficient for modeling contaminated soil-water systems. Researchers are thus faced with the
difficult responsibility of assembling thermodynamic data from other possibly less-credible
publications, borrowing values from extant chemical reaction models, and/or conducting their
own reviews of published thermodynamic data. Because there is a growing reliance on
thermodynamic review efforts completed by coworkers and other research organizations,
documentation supporting these reviews and the rationale for selecting each datum that is
“accepted” for a model's database are extremely important with respect to (1) defining the
credibility of the database, (2) achieving an internally consistent database, (3) minimizing
duplication in future review efforts, and (4) describing the selection criteria and calculation
methods used in selecting the best values.

1
Personal communication from N. T. Loux at the U.S. Environmental Protection Agency in
Athens, Georgia.
5.33
5.2.7.2 Sorption Database
The MINTEQA2 code is not designed to have a thermochemical database, analogous to the
thermodynamic database, that is integrated with the adsorption submodel and its 7 model options.
The adsorption reactions and associated model parameters need to be supplied by the user as part
of each input file. This process and example input files are discussed in Allison et al. (1991).
However, the current MINTEQA2/PRODEFA2 software package is supplied with a limited
adsorption data file for use with the diffuse layer adsorption model option. Data files are not
supplied for any of the other adsorption model options. The data file, formatted in ASCII, is
named FEO-DLM.dbs. It includes surface reactions and associated intrinsic conditional surface
complexation constants applicable to the diffuse layer model for the adsorption of the trace metals
Ba
2+
, Be
2+
, Ca
2+
, Cd
2+
, Cu
2+
, Ni
2+
, Pb
2+
, and Zn
2+
, and the ligands H
3
AsO
3
E (aq), H
3
AsO
4
E (aq),
H
3
BO
3
E (aq), PO
4
3-
, and SO
4
2-
onto 2 types of iron-oxide sites. The adsorption constants are based
of data published by Dzombak (1986).
1
5.2.7.2.1 Status Relative to Project Scope
Of the elements chosen for study in this project, cadmium and lead are the only 2 elements
included in the diffuse layer adsorption model data file supplied with MINTEQA2. As mentioned
above, this file is restricted to adsorption onto 2 types of iron-oxide sites, and is therefore not
applicable for the adsorption of these metals to other mineral reactive surfaces. None of the other
contaminants, including chromium, cesium, plutonium, radon, strontium, thorium, or uranium, are
supported by this data file.
The MINTEQA2/PRODEFA2 software package includes no adsorption database files for the
activity partition coefficient (K
d
act
), activity Langmuir isotherm, activity Freundlich isotherm, ion
exchange, constant capacitance, or triple layer adsorption models.
5.2.7.2.2 Published Database Sources
No published compilations are known to exist for adsorption constants for the activity partition
coefficient (K
d
act
), activity Langmuir isotherm, activity Freundlich isotherm, and ion exchange
adsorption models used in MINTEQA2. Numerous individual data sets have been published for
the adsorption of many individual contaminants on specific mineral substrates, such as TiO
2
or
goethite. Typically, these data are parameterized using 1 or more of the surface complexation
models. Compilations and review of these studies was beyond the scope of this project.
Smith and Jenne (1988) (and related papers by Dzombak and Hayes, 1992, and Smith and Jenne,
1992) compiled and evaluated published values for triple layer model constants for the adsorption
5.34
of numerous constituents on "-FeO(OH), amorphous iron(III) hydrous oxide, and *-MnO
2
solids.
This study was conducted for the U.S. Environmental Protection Agency (Athens, Georgia) for
use in modeling the migration of contaminants in ground-water systems. Their compilation
included intrinsic constants and associated reaction stoichiometries for the adsorption of species
containing the following constituents:
C For adsorption onto Fe(III) hydrous oxides: Ag, As(V), Ba, CO
3
, Ca, Cd, Co, Cr(VI),
Cu(II), Fe(II), Hg(II), Mg, Mn(II), Np(V), Pb, Pu(IV), Sb(III), Sb(V), Se(VI), Se(IV), S,
SO
4
, Th(I), U(VI), and Zn
C For adsorption onto *-MnO
2
: Ag, Ba, Ca, Cd, Co, Cu(II), Fe(II), Hg(II), Mg, Mn(II), Pb,
Th(I), and Zn.
Turner (1995) compiled and critically reviewed adsorption data reported in the literature for
surface complexation models. He then used a uniform approach to parameterize these data using
the diffuse layer, constant capacitance, and triple layer surface complexation models. His study
was conducted in support of research funded by NRC to study the potential migration of
radionuclides associated with the geologic disposal of commercial high level radioactive waste.
Turner (1993) previously described the use of the MINTEQA2 chemical reaction code to model
adsorption of radionuclides. Turner (1995) reported model constants for:
C Americium(III) on "-Al
2
O
3
, (-Al
2
O
3
, and amorphous SiO
2
C Neptunium(V) on "-Al
2
O
3
, (-Al
2
O
3
, boehmite ((-AlOOH), goethite ["-FeO(OH),]
magnetite (Fe
3
O
4
), lepidocrocite [(-FeO(OH)], ferrihydrite (5Fe
2
O
3
·9H
2
O), amorphous
SiO
2
, biotite mica [K(Mg,Fe)
3
(Al,Fe)Si
3
O
10
(OH,F)
2
], and kaolinite [Al
2
Si
2
O
5
(OH)
4
]
C Plutonium(IV) on goethite
C Plutonium(V) on (-Al
2
O
3
and goethite
C Thorium on (-Al
2
O
3
and amorphous SiO
2
C Uranium(VI) on "-Al
2
O
3
, magnetite, ferrihydrite, goethite, quartz (SiO
2
), and kaolinite
C Carbon on ferrihydrite.
We are not aware of any other major published compilations of adsorption thermochemical data
for use with MINTEQA2. Moreover, it is very possible that individual investigators have
compiled and parameterized their own databases of adsorption constants based on the needs of
their individual research projects. General access, especially in these days of cost recovery, and
quality assurance issues will likely prohibit the use of many such individual data files.
5.35
5.3 Adsorption Model Options in MINTEQA2
The MINTEQA2 chemical reaction code includes 7 adsorption model options. Each of these
adsorption models and their associated equations and reactions are briefly described below.
MINTEQA2 includes the following non-electrostatic adsorption models
C Activity partition coefficient (K
d
act
) model
C Activity Langmuir model
C Activity Freundlich model
C Ion exchange model
and electrostatic adsorption models
C Diffuse layer model
C Constant capacitance model
C Triple layer models.
The following descriptions and associated equations are adapted from the MINTEQA2
documentation by Allison et al. (1991). When using the adsorption model options, readers are
cautioned to read the MINTEQA2 documentation carefully relative to correct entry and
formulation of model reactions and associated constants.
It should be noted that the non-electrostatic models in MINTEQA2 are formulated in terms of
species activities, and not the more traditional approach of total concentrations of dissolved metal.
The purpose of this approach is to reduce the dependency of the model parameters to effects from
ionic strength and aqueous complexation of the adsorbing metal.
Limitations remain, however, regarding these activity formulations which restricts the range of
applicability of these non-electrostatic models. These non-electrostatic adsorption models do not
consider: charge balance on surface sites and adsorbed species, electrostatic forces between the
adsorbing species and charged surface of the mineral, and reactions between the mineral and
dissolved constituents other than the adsorbing metal. The effect of these processes changes with
variations in the composition of an aqueous solution. These processes are, however, incorporated
into the more robust, but more data intensive, electrostatic “surface complexation” adsorption
models. The following descriptions of the electrostatic adsorption models incorporated into
MINTEQA2 are cursory. The reader is referred to sources, such as Westall and Hohl (1980),
Morel et al. (1981), Barrow and Bowden (1987), and Davis and Kent (1990), for detailed
descriptions, comparisons, and derivations of the relevant equations and reactions associated with
these models.
5.36
5.3.1 Electrostatic Versus Non-Electrostatic Models
Hydrous oxides of iron, manganese, and aluminum and amorphous aluminosilicates that exist as
discrete mineral grains or surface coatings on other minerals in soils are assumed to be primary
adsorbents for trace metals ions. These solid phases have variable surface charges and exhibit
amphoteric behavior. The solids have a net positive charge at pH values below their point of zero
charge (PZC) and a net negative charge at pH values above the PZC (see Chapter 2).
These surface charges create electrostatic potentials extending into the surrounding solutions.
Dissolved aqueous species that have a charge of the same polarity as the surface will be repelled,
while aqueous species with a charge opposite to that of the surface will be attracted (adsorbed).
The electrostatic potentials associated with charged surfaces may therefore affect the adsorption
of dissolved species on these surfaces. Unlike the non-electrostatic adsorption models, the
electrostatic models include a component that accounts for the electrostatic potentials at the
charged surface. The mass action equations of electrostatic adsorption models include terms that
modify the activities of adsorbing species approaching charged surfaces by the electrical work
necessary to penetrate the zone of electrostatic potentials (R's) associated with the mineral
surface. The 3 electrostatic models in MINTEQA2 differ primarily in the types of surface species
that are allowed within specific physical locations or layers extending away from the surface and
in the parameters that each model uses.
The 3 electrostatic models in MINTEQA2 deal with adsorption as surface complexation reactions
analogous to aqueous complexation reactions in solution. In the descriptions of the MINTEQA2
adsorption models that follow, surface sites are represented in the adsorption reactions and mass
action expressions as SOH groups, where S refers to the mineral structure and adsorption site
located at the solid-liquid interface. Some ions, such as H
+
, OH
-
, and a variety of trace metal ions
are assumed to be adsorbed by complexation with these surface sites.
In the triple layer model (TLM), the most complicated of the 3 electrostatic adsorption models in
MINTEQA2, the space around the solid surface is represented in surface complexation adsorption
models as 3 semi-infinite layers or zones between the solid surface and the solution (Figure 5.4).
These zones are separated by the o, $, and d planes. Starting at the mineral surface, The o plane
represents the first interface between the solid surface and the aqueous phase. Generally, only the
H
+
and OH
-
ions are allowed to penetrate the o layer to interact with the solid surface. Beyond
the o plane, farther from the mineral surface, is the $ plane which ends at the boundary of the
diffuse zone, the d plane. Dissolved ions, such as macro constituents (e.g., Na
+
, Ca
2+
, and SO
4
2-
)
and trace constituents ions that are adsorbing onto the solid surface are allowed into the $ layer.
The third layer is the diffuse zone where the ions are not influenced strongly by electrostatic
charge on the solid surface. The ions in this region are considered to be counterions that
neutralize any residual charge caused by the surface and adsorbed ions in the $ layer. Continuing
further from the mineral surface, the d layer blends into the bulk solution.

5.37
Distance From Surface (x)
Diffuse Layer of
Counter Ions
Schematic of
Surface Species
Schematic of
Charge-Potential
Relationships
Potential
(
ψ
)
σ
o
ψ
o
´O´ Plane: ψ
o
& σ
o
X——O - H
o
X——O - H
2
+
X——O - ( )
-
X——O - H
o
X——O - ( )
-
- Cd
2+
X——O - ( )
-
- CdOH
+
Constants
Corresponding
to Surface Species
K
a2
K
a1
*
K
Cd
*
K
CdOH
´b´ Plane: ψ
β
& σ
β
´d´ Plane: ψ
d
& σ
d
ψ
ψ
β
ψ
d
C
1
C
2
x
X——O - H
o
X——O - ( )
-
- Na
+
X——O - H
2
+
- Cl
-
X——O - H
2
+
- SO
4
2-
*
K
Cl
*
K
Na
*
K
SO4
Figure 5.4. Schematic representation of the triple layer model showing
surface species and surface charge-potential relationships.
[Taken from Peterson et al. (1987a). Brackets in the o
plane indicated deprotonated surface sites.]

5.38
Distance From Surface (x)
Diffuse Layer of Counter Ions
Schematic of
Surface Species
Schematic of
Charge-Potential
Relationships
Potential (ψ)
ψ
o
σ
o
C´
ψ(x)
´d´ Plane or Outer
Helmholtz Plane (OH)
´O´ Plane at Potential ψ
o
and Charge
σ
o
X——O - H
o
X——O - H
2
+
X——O - ( )
-
X——O - H
o
X——O - Cd
2+
X——O
X——O
Constants
Corresponding
to Surface Species
K
a2
K
a1
*
K
Cd1
*
K
Cd2
Cd
The conceptual models for the constant capacitance (Figure 5.5) and diffuse layer models are
simplified to only 2 zones separated by the o and d planes. The difference between these
2 adsorption models is in the function relating total surface charge, T
F
, to surface potential R
o
(discussed in Sections 5.3.6 and 5.3.7). This function [R(x) in Figure 5.5)] is linear and
exponential, respectively, in the constant capacitance and diffuse layer models. It should be noted
that parameters subscripted with “o” in that 2-layer models are not equivalent to the o plane
parameters defined for the triple layer model due to differences in the definition of the o plane.
Figure 5.5. Schematic representation of the constant capacitance layer model showing
surface species and surface charge-potential relationships. [Taken from
Peterson et al. (1987a). Brackets in the o plane indicated deprotonated
surface sites.]
5.39
F % F
d
' 0 .
(5.10)
F
o
% F
$
' F
(5.11)
(F
o
% F
$
) % F
d
' 0 .
(5.12)
{X
z
s
} ' {X
z
}[e
&RF/RT
]
z
(5.13)
In all 3 models, a charge, F, associated with the surface is assumed to be balanced by a charge (F
d
)
associated with the diffuse layer d of counterions such that
In the constant capacitance and diffuse-layer models, all adsorbed ions contribute to the surface
charge. However, the net charge F due to adsorption in the triple layer model is the sum of the
charges associated with 2 rather than 1 adsorbing plane. These include the innermost o plane and
the $ plane, which are characterized by charges F
o
and F
$
, respectively. Thus, for the triple layer
model, the net surface charge is given by
which is balanced by the charge in the diffuse layer such that
Because the electrical potential gradients extending away from the mineral’s surface result from
the surface charge, the specifically adsorbed potential determining ions also govern distributions
of counterions in the diffuse layer.
Activities of ions in solution and near the surface are influenced by the presence of electrostatic
potentials arising from the surface charge. The activity difference between ions near the surface
and those far away is the result of electrical work required to move them across the potential
gradient between the charged surface and the bulk solution. The activity change between these
zones is related to the ion charge, z, and the electrical potential, R, near the surface and can be
expressed using the exponential Boltzmann expression,
where z = charge of ion X,
{X
s
z
} = activity of an ion X of charge z near the surface,
{X
z
} = activity of ion X in bulk solution beyond the influence of the charged surface,
e
-RF/RT
= Boltzmann factor,
F = Faraday constant,
R = ideal gas constant, and
T = absolute temperature in Kelvin.
The general algorithm is similar for all 3 electrostatic models in MINTEQA2. Each model is only
briefly described below. The surface reactions for the electrostatic models in MINTEQA2 are
written with the Boltzmann factor included as an reactant component with a stoichiometric factor
appropriate for the reaction. Although these electrostatically-related components are included in
the mass action equations, they are not analogous to the chemical components defined in

5.40
K
d
'
Amount of element sorbed on solid / solid mass
Amount of element dissolved in solution / solution volume
.
(5.14)
SOH % M XX SOH·M
(5.15)
K
d
'
[SOH·M]
[M]
total diss
(5.16)
MINTEQA2 and have no analytical totals for their input values. Their total charges are
determined from equations that are unique to each electrostatic model and potential. The activity
coefficients for the Boltzmann factor components are set to unity in MINTEQA2.
Adsorption reactions are entered as part of MINTEQA2 input files. The MINTEQA2 code, as
noted previously, has no integrated adsorption database. The adsorption reactions and associated
equilibrium constants are written in terms of the neutral surface site, SOH. They are entered as
formation reactions, analogous to the aqueous complexation and mineral solubility reactions
included in the thermodynamic database. Published adsorption reactions and associated constants
are, however, sometimes referenced to the protonated surface site SOH
2
+
for adsorbing anions and
the deprotonated site SO
-
for adsorbing cations. In these cases, the user must modify the
published reaction and equilibrium constant data in terms of MINTEQA2 components to use them
in a MINTEQA2 input file.
5.3.2 Activity Partition Coefficient (K
d
) Model
The traditional partition coefficient, K
d
, adsorption model (see Chapter 2) is defined as the ratio of
the concentration of metal bound on the surface of the solid to the total concentration of metal
dissolved in the liquid phase at equilibrium as in
This process can be expressed as the surface adsorption reaction
where SOH = unreacted surface site,
M = a dissolved metal M, and
SOH·M = adsorption site occupied by a component or surface-bound metal M.
The convention used for symbols in the adsorption model equations discussed in this chapter
follows that used by Allison et al.(1991). Although the basic adsorption equations are
comparable to those listed in Chapter 2, the symbols may be differ slightly.
The mass action expression for this reaction is
where [SOH·M] = concentration of adsorption sites occupied by a component M or surface-
bound metal per unit mass of adsorbing solid

5.41
K
act
d
'
{SOH·M}
{M}
'
[SOH·M]
(
M
[M]
(5.17)
[M]
total diss
= total concentration of dissolved M at equilibrium.
Following common convention for thermodynamic nomenclature, reaction species indicated
within [ ] refer to concentrations, and those indicated within { } refer to activities. Equation 5.16
assumes that the concentration of unreacted surface sites, SOH, are in great excess relative to the
total concentration of dissolved metal and the activity of SOH is equal to 1.
As mentioned previously, the traditional K
d
assumes that all species of metal M absorb with equal
strength, and [M]
total diss
includes all aqueous species containing metal M. For example, using the
species listed in the MINTEQA2 thermodynamic database, the total concentrations of dissolved
lead, [Pb]
total diss
, in the absence of any organic complexants in the water, could include the
following species:
[Pb]
total diss
= [Pb
2+
] + [PbOH
+
] + [Pb(OH)
2
"
(aq)] + [Pb(OH)
3
-
] +
[Pb
2
OH
3+
] + [Pb
3
(OH)
4
2+
] + [Pb(OH)
4
2-
] +
[PbCO
3
"
(aq)] + [Pb(CO
3
)
2
2-
] + [PbHCO
3
+
] + [PbNO
3
+
] +
[PbSO
4
"
(aq)] + [Pb(SO
4
)
2
2-
] + [Pb(HS)
2
"
(aq)] + [Pb(HS)
3
-
] +
[PbCl
+
] + [PbCl
2
"
(aq)] + [PbCl
3
-
] + [PbCl
3
2-
] +
[PbF
+
] + [PbF
2
"
(aq)] + [PbF
3
-
] + [ PbF
3
2-
] +
[PbBr
+
] + [PbBr
2
"
(aq)] + [PbI
+
] + [PbI
2
"
(aq)].
In the presence of organic complexants, [Pb]
total diss
would also include, in addition to the lead
species listed above, the concentrations of aqueous lead citrate, acetate, EDTA, HEDTA, and
other organic complexes.
Because experimental data suggest that only certain aqueous species react with the surface of a
mineral, the traditional K
d
model is reformulated in MINTEQA2 in terms of the activities of
species to provide the activity K
d
act
model.
In MINTEQA2, the mass action expression for the activity K
d
act
model is
where {M} = free activity of the uncomplexed “bare” cation of M in the equilibrium solution,
(
M
= activity coefficient of dissolved species M
The quantity {SOH·M} is defined as equal to [SOH·M]. This assumption is made because there
is no generally accepted method for calculating activity coefficients for unreacted or reacted
adsorption sites. The parameter K
d
act
can be considered the equilibrium constant for the surface

5.42
[SOH·M] '
K
L
[SOH]
total
[M]
total diss
1 % K
L
[M]
total diss
(5.18)
SOH % M XX SOH·M .
(5.19)
K
act
L
'
{SOH·M}
{M}{SOH}
'
(
SOH·M
[SOH·M]
(
M
[M](
SOH
[SOH]
.
(5.20)
K
act
L
'
[SOH·M]
(
M
[M][SOH]
.
(5.21)
[SOH]
total
' [SOH·M] % [SOH] .
(5.22)
reaction described in Equation 5.15. This model assumes that there is an unlimited supply of
unreacted adsorption sites and the mineral surface cannot become saturated regardless of how
much M adsorbs.
5.3.3 Activity Langmuir Model
The concentration-based Langmuir adsorption model has the constraint that the number of surface
sites available for adsorption is limited. This is the only difference between the Langmuir and K
d
adsorption models. The partition coefficient, K
d
, model is linear with respect to the total
concentration of a dissolved metal, whereas the Langmuir model is non-linear. The user must
specify the concentration of available adsorption sites as part of the input file. The Langmuir
equation for adsorption is defined by
where K
L
= Langmuir adsorption constant,
[SOH·M] = amount of adsorbed metal M per unit mass of adsorbing solid,
[SOH]
total
= total concentration of available surface adsorption sites, and
[M]
total diss
= total concentration of dissolved metal M at equilibrium.
The surface adsorption reaction used for the Langmuir model is identical to that for the K
d
model
The equilibrium constant, K
L
act
, for this reaction can be expressed in terms of activities as
As discussed previously, the activity coefficients pertaining to unreacted and reacted surface sites
in this and the other adsorption models in MINTEQA2 are assigned values of unity.
Equation 5.20 can then be rewritten as
The mass balance equation for the available surface sites is
By combining Equations 5.21 and 5.22 in terms of [SOH]
total
and [SOH·M], one obtains the
Langmuir relationship in terms of activities

5.43
[SOH·M] '
K
act
L
[SOH]
total
(
m
[M]
1 % K
act
L
(
m
[M]
.
(5.23)
SOH % M
1
XX SOH·M
1
K
act
L,1
(5.24)
SOH % M
2
XX SOH·M
2
K
act
L,2
(5.25)
SOH % M
n
XX SOH·M
n
K
act
L,n
(5.26)
[M]
total diss
[SOH·M]
'
1
K
L
[SOH]
total
%
[M]
total diss
[SOH]
total
.
(5.27)
By substituting K
L
act
with K
L
and setting (
m
to a value of 1, Equation 5.23 reduces to the
concentration Langmuir model expressed in Equation 5.18.
To use MINTEQA2 to model competition between different metals for adsorption on the
available surface sites, one must define the separate adsorption reactions on the surface. For the
competitive Langmuir model for the competing metals M
1
, M
2
,... M
n
, separate reactions with
associated mass balance expressions need to be formulated using Equations 5.19-5.21 such that
·
·
Geochemical modeling and plotting techniques may be used to derive constants for the activity
Langmuir model from experimentally-measured, concentration-based K
L
data. One must first
determine if the concentration-based Langmuir model fits the experimental data by using the linear
form of Equation 5.18
A plot of [M]
total diss
/[SOH·M] versus [M]
total diss
will result in a straight line with the slope
1/[SOH]
total
and intercept 1/K
L
[SOH]
total
if the data fit the Langmuir isotherm. The value for the
concentration-based K
L
is obtained by dividing this slope by the intercept. Geochemical modeling
is then used to calculate the aqueous speciation of metal M for the composition of the aqueous
solution in which the K
L
data were determined. The K
L
act
value can then be derived from an
analogous plot in which the calculated activities {M} for metal M are plotted in place of the
concentration term [M].

5.44
[SOH·M] ' K
F
[M]
total diss
1/N
.
(5.28)
SOH %
1
N
M XX SOH·M .
(5.29)
K
act
F
'
{SOH·M}
{M}
1/N
{SOH}
.
(5.30)
K
act
F
'
[SOH·M]
{M}
1/N
(5.31)
5.3.4 Activity Freundlich Model
The concentration-based Freundlich equation for adsorption is defined by
where K
F
= Freundlich adsorption constant,
[SOH·M] = amount of adsorbed metal M per unit mass of adsorbing solid,
[M]
total diss
= total concentration of dissolved metal M at equilibrium, and
N = a constant.
The Freundlich equation is sometimes written with the exponent in Equation 5.28 being N instead
of 1/N. The Freundlich model assumes, like the K
d
adsorption model, an unlimited supply of
unreacted adsorption sites. For the special case where N equals 1, the mass action equations for
the Freundlich and K
d
models are identical.
The Freundlich model can be considered as a surface adsorption reaction where the stoichiometric
coefficient for the adsorbed metal M equals 1/N as in
The equilibrium constant, K
F
act
, for this reaction can be expressed in terms of activities as
Like the activity K
d
act
model, there is no mass balance on surface sites, and, assuming an excess of
sites with respect to adsorbed metal M, the concentration, [SOH], and activity {SOH}, of the
unreacted surface sites are assumed equal and set to 1. Under these conditions and assuming
{SOH·M} equals [SOH·M] as with the activity K
d
act
and Langmuir models, Equation 5.30
becomes
which is similar to the K
d
act
model except that the stoichiometric coefficient 1/N of the adsorbing
species of metal M.
An approach using geochemical modeling and plotting techniques similar to that described for the
activity Langmuir model may be used to calculate constants for the activity Freundlich model
from experimentally-measured, concentration-based K
F
data. One must first determine if the
Freundlich model fits the experimental data by using the logarithmic form of the Freundlich mass
action Equation 5.28

5.45
log [SOH·M] ' log K
F
%
1
N
log [M]
total diss
.
(5.32)
a SOH·M
1
% b M
2
' b SOH·M
2
% a M
1
.
(5.33)
K
ex
'
{M
1
}
a
{SOH·M
2
}
b
{M
2
}
b
{SOH·M
1
}
a
'
(
a
M
1
[M
1
]
a
[SOH·M
2
]
b
(
b
M
2
[M
2
]
b
[SOH·M
1
]
a
.
(5.34)
T
F
o
' 0.1174 I
½
sinh(ZR
o
F/2RT)
(5.35)
If the data fit the model, a plot of log [SOH·M] versus log [M]
total diss
will result in a straight line
with the slope 1/N and intercept log K
F
. Geochemical modeling is then used to calculate the
aqueous speciation of metal M for the composition of the aqueous solution in which the K
F
data
were determined. The K
F
act
value can then be derived by plotting the calculated activities {M} for
the adsorbing species of metal M in place of the concentration term [M]
total diss
.
5.3.5 Ion Exchange Model
Ion exchange sorption is defined as the process by which a dissolved ion M
2
is exchanged for an
ion M
1
that already occupies a surface sorption site and ion M
1
is in turn released back into
solution. The ion exchange reaction can be expressed as
where M
1
= the ion initially occupying the exchange site,
M
2
= the ion replacing M
1
on the exchange site;
SOH·M
1
= surface sites occupied by ion M
1
SOH·M
2
= surface sites occupied by ion M
2
, and
a and b = stoichiometric coefficients.
The equilibrium constant (selectivity coefficient), K
ex
, for the exchange reaction expressed as
The constant K
ex
can be written in terms of concentrations by replacing activity of each species
with the product of concentration and activity coefficient. The activity coefficients for the
occupied sites, SOH·M
n
, are set equal to one as was assumed for the previous adsorption models
in MINTEQA2.
5.3.6 Diffuse Layer Model
For the diffuse-layer model, the total charge, T
F
, for plane o is calculated as
where Z = valency of the symmetrical electrolyte (which we take as unity),
I = ionic strength, and

5.46
SOH % H
%
s
X SOH
%
2
(5.36)
K '
{SOH
%
2
}
{SOH}{H
%
s
}
(5.37)
{H
%
s
} ' {H
%
}e
&R
o
F/RT
(5.38)
SOH % H
%
% e
&R
o
F/RT
X SOH
%
2
(5.39)
K '
{SOH
%
2
}
{SOH}{H
%
}[e
&R
o
F/RT
]
(5.40)
SOH & H
%
s
X SO
&
(5.41)
SOH & H
%
& e
&R
o
F/RT
X SO
&
(5.42)
all other parameters are defined as in Equation 5.13.
Examples of surface reactions are listed below for protonation and deprotonation reactions as
well as for a divalent cation M
2+
. Boltzmann factors are represented in the mass action as
components.
The surface reaction and corresponding mass action expression for the protonation reaction are,
respectively,
and
where H
s
+
denotes a hydronium ion near the surface.
The activity coefficients for the surface species SOH
2
+
and SOH are assumed to be equal to unity.
The activity of H
s
+
must be corrected for the energy change required to move from the bulk
solution to the charged surface. This activity change is represented by expressing {H
s
+
} in terms
of the activity of the bulk solution hydronium ion {H
+
} and associated exponential Boltzmann
expression for a charge z of 1 as
Substituting this expression for {H
s
+
} in Equations 5.36 and 5.37, one obtains the following
surface reaction and mass action equation expressed in terms of the Boltzmann factor
and
The stoichiometry for the corresponding de-protonation reaction is
Substituting for {H
s
+
} as above results in the following de-protonation surface reaction and mass
action equation

5.47
K '
{SO
&
}{H
%
}[e
&R
o
F/RT
]
{SOH}
. (5.43)
SOH % M
2%
s
& H
%
s
X SO·M
%
.
(5.44)
K '
{SO·M
%
}{H
%
s
}
{SOH}{M
2%
s
}
'
{SO·M
%
}{H
%
}[e
&R
o
F/RT
]
{SOH}{M
2%
}[e
&R
o
F/RT
]
2
(5.45)
K '
{SO·M
%
}{H
%
}
{SOH}{M
2%
}[e
&R
o
F/RT
]
(5.46)
T
F
o
. C R
o
(5.47)
and
The stoichiometry for a surface reaction involving a multivalent species, such as a divalent cation
M
2+
, is
The mass action expression for this type of adsorption reaction also includes the charge and
stoichiometry for the adsorbing ion. Substituting for {M
s
2+
} and for {H
s
+
} in Equation 5.44, one
obtains the following mass action expressions
Mass action expressions for other surface reactions are formulated in a similar manner.
5.3.7 Constant Capacitance Model
The constant capacitance model is a special case of the diffuse layer model, applicable in theory
only to systems at high, constant ionic strength. The constant capacitance model is similar to the
diffuse layer model in that they both define specific adsorption of all ions on the o plane. Except
for the values of the equilibrium constants, the mass action and charge balance equations are
identical for the these 2 adsorption models. Therefore, the surface reactions and mass action
expressions described above for the diffuse layer model also apply to the constant capacitance
model.
The difference in these 2 models is in the function relating total surface charge, T
F
, to surface
potential R
o
. In the constant capacitance model, Equation 5.35 is approximated by
where C is a constant capacitance term. Although the constant capacitance and diffuse layer
models are implemented similarly, the capacitance term C is often treated as a fitting parameter
rather than as a measured characteristic of the system.
5.48
T
F
o
' C
1
(R
o
& R
$
)
(5.48)
T
F
$
' C
1
(R
$
& R
o
) % C
2
(R
$
& R
d
)
(5.49)
T
F
d
' C
2
(R
d
& R
$
)
(5.50)
SOH & H
%
s
% M
%
s
X (SO·M) .
(5.51)
5.3.8 Triple Layer Model
The triple layer model (Figure 5.4) includes 2 adsorbing planes instead of 1 plane as
conceptualized in the diffuse layer and constant capacitances models. As implemented in
MINTEQA2, the o plane, the inner most zone, only includes the protonation and deprotonation
(i.e., gain or loss of H
+
) reactions at the surface sites. The $ plane includes other specifically
adsorbed ions with charge F
$
and potential R
$
in that zone. The diffuse layer or 'd' plane, which is
the outer most zone, includes non-specifically adsorbed ions affected by R
d
potentials. The
capacitances between the o and $ planes and the $ and d planes are designated C
1
and C
2
,
respectively. The user must provide values for both capacitance terms.
The total charges, T
F
o
, T
F
$
, and T
F
d
, associated with o, $, and d planes, respectively, in the triple-
layer model are defined as
where R
o
= electrostatic potential at the o plane,
R
$
= electrostatic potential at the $ plane, and
R
d
= electrostatic potential at the d plane.
Surface reactions as expressed in the triple layer model differ from those used for the diffuse layer
and constant capacitance models only in that their mass action expressions include the proper
stoichiometry for the electrostatic components representing the $ and o planes. The d plane,
which as no specific adsorption, is therefore not a factor in the stoichiometry.
The surface protonation and deprotonation reactions for the triple layer model, except for their
associated equilibrium constant values, are identical to those given above for the diffuse layer
models. Examples of surface reactions and mass action expressions for the adsorption of a mono-
and divalent cations and a monovalent anion adapted from Allison et al. (1991) are given below
for the triple layer model. They show the stoichiometric coefficients for the electrostatic
components representing the $ and o planes.
The surface reaction for the adsorption of the monovalent metal cation M
+
is

5.49
{H
%
s
} ' {H
%
} [e
&R
o
F/RT
] (5.52)
{M
%
s
} ' {M
%
} [e
&R
$
F/RT
] . (5.53)
SOM & H
%
& e
&R
o
F/RT
% M
%
% e
&R
$
F/RT
X SO·M
(5.54)
K '
{SO·M}{H
%
}[e
&R
o
F/RT
]
{SOH}{M
%
}[e
&R
$
F/RT
]
.
(5.55)
SOH & H
%
s
% M
2%
s
X (SO·M)
%
.
(5.56)
{M
2%
s
} ' {M
2%
}[e
&R
$
F/RT
]
2
(5.57)
SOH & H
%
& e
&R
o
F/RT
% M
2%
% 2e
&R
$
F/RT
X SO·M
%
(5.58)
K '
{SO·M
%
}{H
%
}[e
&R
o
F/RT
]
{SOH}{M
2%
}[e
&R
$
F/RT
]
2
.
(5.59)
SOH % A
&
s
% H
%
s
X SOH
2
·A .
(5.60)
In the triple layer model, H
s
+
and M
s
+
occur in the o and $ planes, respectively. Therefore,
and
Substituting these expressions into Equation 5.51, the following MINTEQA2 reaction and mass
action expression are obtained
and
The surface reaction for the adsorption of the divalent metal cation M
2+
is
For the divalent cation adsorbed in the $ plane,
Substituting this expression in the reaction above gives the following MINTEQA2 reaction and
mass action expression
and
The surface reaction for the adsorption of the monovalent anion A
-
is
This reaction results in the formation of a neutral surface complex. For the anion adsorbed in the
$ plane

5.50
{A
&
s
} ' {A
&
}[e
&R
$
F/RT
]
&
. (5.61)
SOH % A
&
& e
&R
$
F/RT
% H
%
% e
&R
o
F/RT
X SOH
2
·A (5.62)
K '
{SOH
2
·A}[e
&R
$
F/RT
]
{SOH}{A
&
}{H
%
}[e
&R
o
F/RT
]
.
(5.63)
Substituting this into the above anion adsorption reaction, one obtains the following MINTEQA2
reaction and mass action expression
and
The formulation of reactions and mass action expressions for other adsorbing cations and anions
is similar to those examples given above.
5.4 Summary
Chemical reaction models are valuable computational tools that may be used to analyze the
macro-chemical processes (e.g., aqueous complexation, redox, solubility, and adsorption
equilibrium) affecting the composition of a soil-water system being studied in the laboratory, field
lysimeter, or field site. They also be used to provide some bounding calculations for predicting
the changes in chemistry that will result when 1 or more of these processes are imposed on a soil-
water system.
Numerous chemical reaction models exist. The MINTEQA2 computer code was developed with
EPA funding and is currently distributed by EPA in a form that executes on personal computers.
MINTEQA2 includes aqueous speciation, solubility (i.e., saturation indices),
precipitation/dissolution, and adsorption submodels. MINTEQA2's adsorption submodel includes
4 non-electrostatic [activity partition coefficient (K
d
act
), activity Langmuir, activity Freundlich, and
ion exchange] models and 3 electrostatic (diffuse layer, constant capacitance, and triple layer)
adsorption model options.
MINTEQA2 and other similar chemical reaction models can be used in indirect ways to support
evaluations of K
d
values and related contaminant migration and risk assessment modeling. These
applications include the following:
C Calculation of aqueous speciation to determine the ionic state and composition of the
dominant species for a dissolved contaminant present in a soil-water system
C Calculation of bounding, technically-defensible maximum concentration limits for
contaminants (based on solubility constraints) as a function of key composition parameters
(e.g., pH) of any specific soil-water system
5.51
C Analysis of data from laboratory measurements of K
d
values to determined if any solubility
limits were exceeded during the experiments.
Chemical reaction models, however, cannot be used to predict a K
d
value. The user must supply
the adsorption parameters when using any of the adsorption model options. However,
MINTEQA2 may be used to predict the chemical changes that result in the aqueous phase from
adsorption using any of 7 adsorption model options.
The MINTEQA2 model includes an extensive thermodynamic database that is integrated with the
aqueous speciation, solubility, and precipitation/dissolution submodels. Of the elements included
in the project scope, the thermodynamic database distributed by EPA with MINTEQA2 does not
contain reactions and associated thermodynamic data for aqueous species and solids containing
cesium, plutonium, radon, and thorium. Published compilations of thermodynamic data for
aqueous species, solids, and gases containing these elements are available that can be used as
starting points for upgrading the MINTEQA2 database to include cesium, plutonium, radon, and
thorium aqueous species and solids. MINTEQA2 does not have per se an integrated adsorption
submodel database. The adsorption reactions and associated model parameters must be supplied
by the user as part of each input file.
6.1
6.0 REFERENCES
Alberty, R. A. 1987. Physical Chemistry. Seventh Edition, John Wiley and Sons, New York,
New York.
Allison, J. D., D. S. Brown, and K. J. Novo-Gradac. 1991. MINTEQA2/PRODEFA2, A
Geochemical Assessment Model for Environmental Systems: Version 3.0 User's Manual.
EPA/600/3-91/021, U.S. Environmental Protection Agency, Athens, Georgia, March 1991.
Ames, L. L., and J. E. McGarrah. 1980. Basalt Radionuclide Distribution Coefficient
Determinations, FY1979 Annual Report. PNL-3146, Pacific Northwest Laboratory,
Richland, Washington.
Ames, L. L., J. E. McGarrah, B. A. Walker, and P. F. Salter. 1982. “ Sorption of Uranium and
Cesium by Hanford Basalts and Associated Secondary Smectite.” Chemical Geology,
35:205-225.
Ames, L. L., and D. Rai. 1978. Radionuclide Interactions with Soil and Rock Media. EPA
520/6-78-007, U.S. Environmental Protection Agency, Office of Radiation Programs, Las
Vegas, Nevada.
ASTM (American Society of Testing and Materials). 1987. “24-hour Batch-Type Measurement
of Contaminant Sorption by Soils and Sediments.” In Annual Book of ASTM Standards,
Water and Environmental Technology, Volume 11.04, pp. 163-167, Philadelphia,
Pennsylvania.
ASTM (American Society of Testing and Materials). 1988. “Determining a Sorption Constant
(K
oc
) for an Organic Chemical in Soil and Sediments.” In Annual Book of ASTM Standards,
Water and Environmental Technology. Volume 11.04, pp. 731-737, Philadelphia,
Pennsylvania.
Atkinson, A. 1983. “Mathematical Modeling of Leaching From Porous Nuclear Waste-Forms.”
Radioactive Waste Management and the Nuclear Fuel Cycle, 3:371-386.
Atkinson, A., K. Nelson, and T. M. Valentine. 1986. “Leach Test Characterization of Cement-
Based Nuclear Waste Forms.” Nuclear and Chemical Waste Management, 6:241-253.
Atkinson, A., and A. K. Nickerson. 1988. “Diffusion and Sorption of Cesium, Strontium, and
Iodine in Water-Saturated Cement.” Nuclear Technology, 81:100-113.
6.2
Backhus, D. A., J. N. Ryan, D. M. Groher, J. K. MacFarlane, and P. M. Gschwend. 1993.
“Sampling Colloids and Colloid-Associated Contaminants in Ground Water.” Ground Water,
31:466-479.
Baetslé, L. H. 1967. “Migration of Radionuclides in Porous Media.” In Progress in Nuclear
Energy, Series XII, Health Physics, A.M.F. Duhamel (ed.), pp. 707-730, Pergamon Press,
Elmsford, New York.
Ball, J. W., E. A. Jenne, and M. W. Cantrell. 1981a. WATEQ3: A Geochemical Model with
Uranium Added. Open-File Report 81-1183, U.S. Geological Survey, Menlo Park,
California.
Ball, J. W., and D. K. Nordstrom. 1998. “Critical Evaluation and Selection of Standard State
Thermodynamic Properties for Chromium Metal and Its Aqueous Ions, Hydrolysis Species,
Oxides, and Hydroxides.” Journal of Chemical and Engineering Data, 43:895-918.
Ball, J. W., D. K. Nordstrom, and E. A. Jenne. 1981b. Additional and Revised Thermochemical
Data and Computer Code for WATEQ2--A Computerized Chemical Model for Trace and
Major Element Speciation and Mineral Equilibria of Natural Waters. Water Resources
Investigations WRI 78-116 (second printing), U.S. Geological Survey, Menlo Park,
California.
Ball, W. P., C. H. Buehler, T. C. Harmon, D. M. Mackay, and P. V. Roberts. 1990.
“Characterization of a Sandy Aquifer Material at the Grain Scale.” Journal of Contaminant
Hydrology, 5:253-295.
Barber, L. B., II., E. M. Thurman, and D. D. Runnells. 1992. “Geochemical Heterogeneity in a
Sand and Gravel Aquifer: Effect of Sediment Mineralogy and Particle Size on the Sorption of
Chlorobenzenes.” Journal of Contaminant Hydrology, 9:35-54.
Barrow, N. J., and J. W. Bowden. 1987. “A Comparison of Models for Describing the
Adsorption of Anions on a Variable Charge Mineral Surface.” Journal of Colloid and
Interface Science, 119:236-250.
Bates, R. L., and J. A. Jackson (eds.). 1980. Glossary of Geology. American Geological
Institute, Falls Church, Virginia.
Berry, J. A. 1992a. A Review of Sorption of Radionuclides Under the Near- and Far-Field
Conditions of an Underground Radioactive Waste Repository. Part I.
DoE/HMIP/RR/92/061 (Parts I). Harwell Laboratory, Oxfordshire, United Kingdom.
6.3
Berry, J. A. 1992b. A Review of Sorption of Radionuclides Under the Near- and Far-Field
Conditions of an Underground Radioactive Waste Repository. Part II.
DoE/HMIP/RR/92/061 (Parts II). Harwell Laboratory, Oxfordshire, United Kingdom.
Berry, J. A. 1992c. A Review of Sorption of Radionuclides Under the Near- and Far-Field
Conditions of an Underground Radioactive Waste Repository. Part III.
DoE/HMIP/RR/92/061 (Parts III). Harwell Laboratory, Oxfordshire, United Kingdom.
Biggar, J. W., and D. R. Nielsen. 1962. “Miscible Displacement: II. Behavior of Tracers.” Soil
Science Society of America Journal, 26:125-128.
Bolt, G. H., and M. G. M. Bruggenwert (eds.). 1978. Soil Chemistry. A. Basic Elements.
Developments in Soil Science 5A, Elsevier Scientific Publishing Company, New York, New
York.
Bond, W. J., and P. J. Wierenga. 1990. “Immobile Water During Solute Transport in
Unsaturated Sand Columns.” Water Resources Research, 26:2475-2481.
Bosma, W. J. P., and S. E. A. T. Van der Zee. 1993. “Transport of Reacting Solute in a One-
dimensional Chemically Heterogeneous Porous Medium.” Water Resources Research,
29:117-131.
Bosma, W. J. P., S. E. A. T. Van der Zee, and C. J. Van Duijn. 1996. “Plume Development of a
Nonlinearly Adsorbing Solute in Heterogeneous Porous Formations.” Water Resources
Research, 32:1569-1584.
Bourcier, W. L. 1990. Geochemical Modeling of Radionuclide Waste Glass Dissolution Using
EQ3/6: Preliminary Results and Data Needs. UCID-21869, Lawrence Livermore National
Laboratory, Livermore, California.
Bouwer, J. 1991. “Simple Derivation of the Retardation Equation and Application to
Preferential Flow and Macrodispersion.” Ground Water, 29:41-46.
Box, G. E. P., and D. W. Behnken. 1960. “Some New Three Level Designs for the Study of
Quantitative Variables.” Technometrics, 2:455-475.
Brown, P. L., A. Haworth, S. M. Sharland, and C. J. Tweed. 1991. HARPHRQ: A Geochemical
Speciation Program Based on PHREEQE. Report NSS/R188, Harwell Laboratory, Harwell
Laboratory, Oxfordshire, United Kingdom.
Brown, D. S., and J. D. Allison. 1987. MINTEQA1, an Equilibrium Metal Speciation Model:
User's Manual. EPA/600/3-87/012, U.S. Environmental Protection Agency, Athens,
Georgia.
6.4
Brusseau, M. L. 1994. “Transport of Reactive Contaminants in Heterogeneous Porous Media.”
Reviews of Geophysics, 32:285-313.
Brusseau, M. L., and P. S. C. Rao. 1989. “Sorption Nonideality During Organic Contaminant
Transport in Porous Media.” CRC Critical Reviews in Environmental Control, 19:33-99.
Brusseau, M. L., and J. M. Zachara. 1993. “Transport of Co
2+
in a Physically and Chemically
Heterogeneous Porous Media.” Environmental Science and Technology, 27:1937-1939.
Buck, J. W., G. Whelan, J. G. Droppo, Jr., D. L. Strenge, K. J. Castleton, J. P. McDonald,
C. Sato, and G. P. Streile. 1995. Multimedia Environmental Pollutant Assessment System
(MEPAS): Application Guidance -- Guidelines for Evaluating MEPAS Input Parameters for
Version 3.1. PNL-10395, Pacific Northwest National Laboratory, Richland, Washington.
Buddemeir, R. W., and J. R. Hunt. 1988. “Transport of Colloidal Contaminants in Groundwater:
Radionuclide Migration at the Nevada Test Site.” Applied Geochemistry, 3:535-548.
Buffle, J., D. Perret, and M. Newman. 1992. “The Use of Filtration and Ultrafiltration for Size
Fractionation of Aquatic Particles, Colloids, and Macromolecules.” In Environmental
Particles. Part II. Sampling and Characterization of Particles of Aquatic Systems, J. Buffle
and H. Van Leeuwen (eds.), pp. 171-230, Lewis Publishers, Chelsea, Michigan.
Burr, D. T., E. A. Sudicky, and R. L. Naff. 1994. “Nonreactive and Reactive Solute Transport in
Three-dimensional Heterogeneous Porous Media: Mean Displacement, Plume Spreading, and
Uncertainty.” Water Resources Research, 30:791-815.
Carter, S. L., M. M. Mortland, and W. D. Kemper. 1986. “Specific Surface.” In Methods of
Soil Analysis -- Part 1: Physical and Mineralogical Methods, A. Klute (ed.), Second
Edition, pp. 413-422, Soil Science Society of America, Madison, Wisconsin.
Carslaw, H. S., and J. C. Jaeger. 1959. Conduction of Heat in Solids. Oxford University Press,
London, England.
Chiou, Cary T., Susan E. McGroddy, and Daniel E. Kile. 1998. “Partition Characteristics of
Polycyclic Aromatic Hydrocarbons on Soils and Sediments.” Environmental Science and
Technology, 32:264-269
Cloninger, M. O., and C. R. Cole. 1981. Reference Analysis on the Use of Engineered Barriers
for Isolation of Spent Nuclear Fuel in Granite and Basalt. PNL-3530, Pacific Northwest
Laboratory, Richland, Washington.
6.5
Cloninger, M. O., C. R. Cole, and J. F. Wahburn. 1980. An Analysis on the Use of Engineered
Barriers for Geologic Isolation of Spent Fuel in a Referenced Salt Repository. PNL-3356,
Pacific Northwest Laboratory, Richland, Washington.
Coats, K. H., and B. D. Smith. 1964. “Dead-End Pore Volume and Dispersion in Porous
Media.” Society of Petroleum Engineers Journal, 4:73-84.
Cochran, G. W., and G. M. Cox. 1957. Experimental Design. Second Edition, Wiley, New
York, New York.
Codell, R. B., K. T. Key, and G. Whelan. 1982. A Collection of Mathematical Models for
Dispersion in Surface Water and Groundwater. NUREG-0868, U.S. Nuclear Regulatory
Commission, Washington, DC.
Cole, C. R., S. B. Yabusaki, and C. T. Kincaid. 1988. CFEST-SC -- Coupled Fluid, Energy, and
Solute Transport Code, SuperComputer Version. Battelle, Pacific Northwest Laboratory,
Richland, Washington.
Corapcioglu, M. Y., and S. Kim. 1995. “Modeling Facilitated Contaminant Transport of Mobile
Bacteria.” Water Resource Research, 31:2639-2647.
Cotton, F. A., and G. Wilkinson. 1972. Advanced Inorganic Chemistry, A Comprehensive Test.
Third Edition. John Wiley and Sons, Inc., New York.
Dagan, G. 1984. “Solute Transport in Heterogeneous Porous Formations.” Journal of Fluid
Mechanics, 145:151-177.
Danielsson, L. G. 1982. “On the Use of Filters for Distinguishing Between Dissolved and
Particulate Fractions in Natural Waters.” Water Research, 16:179-182.
Davies, O. L. 1954. Design and Analysis of Industrial Experiments. Hafner, New York,
New York.
Davis, J. A., and D. B. Kent. 1990. “Surface Complexation Modeling in Aqueous
Geochemistry.” In Mineral-Water Interface Geochemistry, M. F. Hochella, Jr. and
A. F. White (eds.), pp. 177-260, Reviews in Mineralogy, Volume 23, Mineralogical Society of
America, Washington, D.C.
Davis, A., and D. D. Runnells. 1987. “Geochemical Interactions Between Acidic Tails Fluid and
Bedrock: Use of the Computer Model MINTEQ.” Applied Geochemistry, 2:231-241.
6.6
Dayal, R., H. Arora, and N. Morcos. 1983. Estimation of Cesium-137 Release from
Waste/Cement Composites Using Data from Small-Scale Specimen. NUREG/CR-3382,
Nuclear Regulatory Commission, Washington, D.C.
Dean, J. A. (ed.). 1973. Lange’s Handbook of Chemistry. McGraw-Hill, New York, New
York.
Degueldre, C., G. Longworth, V. Mowlin, and P. Vilks. 1989. Grimsel Colloid Exercise. An
International Intercomparison Exercise on the Sampling and Characterization of
Groundwater Colloids. Technical Report 39, Paul Scherrer Institut-Bericht, Wurenlingen und
Villigen, Switzerland.
Delany, J. M. 1985. Reaction of Topopah Spring Tuff with J-13 Water: A Geochemical
Modeling Approach Using the EQ3/6 Reaction Path Code. UCRL-53631 DE86 013226,
Lawrence Livermore National Laboratory, Livermore, California.
Delegard, C. H., and G. S. Barney. 1983. Effects of Hanford High-Level Waste Compounds on
Sorption of Cobalt, Strontium, Neptunium, Plutonium, and Americium of Hanford Sediments.
RHO-RE-ST-1 P, Rockwell Hanford Operations, Richland, Washington.
Delegard, C. H., S. A. Gallagher, and R. B. Kasper. 1981. Saturated Column Leach Studies:
Hanford 216Z-Z-1A Sediment. RHO-SA-210, Rockwell Hanford Operations, Richland,
Washington.
Douglas, L. A. 1989. “Vermiculites.” In Minerals in Soil Environments, J. B. Dixon and
S. B. Week (eds.), Second Edition. pp. 635-674, Soil Science Society of America, Madison,
Wisconsin.
Dragun, J. 1988. Soil Chemistry of Hazardous Materials. Hazardous Materials Control
Research Institute, Silver Spring, Maryland.
Dubinin, M. M., and L. V. Radushkevich. 1947. “Equation of the Characteristic Curve of
Activated Charcoal.” Proceedings of the Academy of Sciences, Physical Chemistry Section,
U.S.S.R., 55:331-333.
Dzombak, D. A. 1986. Toward a Uniform Model for the Sorption of Inorganic Ions on Hydrous
Oxides. Ph.D. Dissertation, Massachusetts Institute of Technology, Boston, Massachusetts.
Dzombak, D. A., and K. F. Hayes. 1992. “Comment on “Recalculation, Evaluation, and
Prediction of Surface Complexation Constants for Metal Adsorption on Iron and Manganese
Oxides.”” Environmental Science and Technology, 26:1251-1253.
6.7
Elabd, H., and W. A. Jury. 1986. “Spatial Variability of Pesticide Adsorption Parameters.”
Environmental Science and Technology, 20:256-260.
EPA (U.S. Environmental Protection Agency). 1984. Uncontrolled Hazardous Waste Site
Ranking System, A User’s Manual. HW-10, U.S. Environmental Protection Agency,
Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1988. Superfund Exposure Assessment Manual.
EPA/540/1-88/001, U.S. Environmental Protection Agency, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1989. Transport and Fate of Contaminants in
the Subsurface. EPA/625/4-89/019, U.S. Environmental Protection Agency, Cincinnati,
Ohio.
EPA (U.S. Environmental Protection Agency). 1991. Site Characterization for Subsurface
Remediation. EPA/625/4-91/026, Office of Research and Development, U.S. Environmental
Protection Agency, Cincinnati, Ohio.
EPA (U.S. Environmental Protection Agency). 1992a. Background Document for Finite Source
Methodology for Wastes Containing Metal. HWEP-S0040, U.S. Environmental Protection
Agency, Office of Solid Waste, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1992b. “U.S. Environmental Protection Agency,
40 CFR 300, Appendix A, Uncontrolled Hazardous Waste Site Ranking System: A User’s
Manual.” Federal Register, 47, No. 137, July 16, 1982.
EPA (U.S. Environmental Protection Agency). 1993. “Environmental Characteristics of EPA,
NRC, and DOE Sites Contaminated with Radioactive Substances.” EPA 402-R-93-011, U.S.
Environmental Protection Agency, Office of Radiation and Indoor Air (6603J), Office of Solid
Waste and Emergency Response, Washington, D.C.
EPA (U.S. Environmental Protection Agency). 1996. Soil Screening Guidance: Technical
Background Document. EPA/540/R-96/018, U.S. Environmental Protection Agency,
Washington, D.C.
EPRI (Electric Power Research Institute). 1991. Use of Batch and Column Methodologies to
Assess Utility Waste Leaching and Subsurface Chemical Attenuation. EPRI EN-7313,
Electric Power Research Institute, Palo Alto, California.
Erdal, B. R., Chairman. 1985. Workshop on Fundamental Geochemistry Needs for Nuclear
Waste Isolation. US DOE CONF8406134, National Technical Information Service,
Springfield, Virginia.
6.8
Felmy, A. R., D. C. Girvin, and E. A. Jenne. 1984. MINTEQ: A Computer Program for
Calculating Aqueous Geochemical Equilibria. EPA-600/3-84-032 (NTIS PB84-157148),
Pacific Northwest Laboratory, Richland, Washington.
Fischer, H. B., E. J. List, R. C.Y. Koh, J. Imberger, and N. H. Brooks. 1979. Mixing in Inland
and Coastal Waters. Academic Press, New York, New York.
Freeze, R. A., and J. A. Cherry. 1979. Groundwater. Prentice-Hall, Englewood Cliffs, New
Jersey.
Friedlander, G., J. W. Kennedy, and J. M. Miller. 1966. Nuclear and Radiochemistry. Second
Edition. John Wiley and Sons, Inc., New York, New York.
Freundlich, H. 1926. Colloid and Capillary Chemistry. Methuen, London, England.
Gamerdinger, A. P., D. I. Kaplan, and C. T. Resch. 1998. Uranium(VI) Sorption and Transport
in Unsaturated, Subsurface Hanford Site Sediments – Effect of Moisture Content and
Sediment Texture. PNNL-11975, Pacific Northwest National Laboratory, Richland,
Washington.
Gamerdinger, A. P., and D. I. Kaplan. 1999. “Application of a Continuous-Flow Centrifugation
Method and Determination of Heterogeneous Solute Transport in Disturbed, Unsaturated
Sediments.” Water Resources Research, (in review).
Garrels, R. M., and C. L. Christ. 1965. Solutions, Minerals, and Equilibria. Freeman, Cooper,
and Company, San Francisco, California.
Gaudet, J. P., H. Jegat, G. Vachaud, and P. J. Wierenga. 1977. “Solute Transfer, with Exchange
Between Mobile and Stagnant Water, Through Unsaturated Sand.” Soil Science Society of
America Journal, 41:665-671.
Gee, G. W., and A. C. Campbell. 1980. Monitoring and Physical Characterization of
Unsaturated Zone Transport - Laboratory Analysis. PNL-3304, Pacific Northwest
Laboratory, Richland, Washington.
Gee, G. W., A. C. Campbell, P. J. Wierenga, and T. L. Jones. 1981. Unsaturated Moisture and
Radionuclide Transport: Laboratory Analysis and Modeling. PNL-3616, Pacific Northwest
Laboratory, Richland, Washington.
Gelhar, L. W., and C. L. Axness. 1983. “Three-Dimensional Stochastic Analysis of
Macrodispersion in Aquifers.” Water Resources Research, 19:161-180.
6.9
Giles, C. H., D. Smith, and A. Huitson. 1974. “A General Treatment and Classification of the
Solute Adsorption Isotherm. I. Theoretical.” Journal of Colloid and Interface Science,
47:755-765.
Glasstone, S. 1972. Thermodynamics for Chemists. Robert E. Krieger Publishing Company,
Huntington, New York.
Goldberg, S. 1995. “Adsorption Models Incorporated into Chemical Equilibrium Models.” In
Chemical Equilibrium and Reaction Models, R. H. Loeppert, A. P. Schwab, and S. Goldberg
(eds.), pp. 75-95, Special Publication Number 42, Soil Science Society of America, Inc.,
Madison, Wisconsin.
Goltz, M. N., and P. V. Roberts. 1986. “Interpreting Organic Solute Transport Data from a
Field Experiment Using Physical Nonequilibrium Models.” Journal of Contaminant
Hydrology,1:77-93.
Grim, R. E. 1968. Clay Mineralogy. McGraw-Hill Book Company, New York, New York.
Gschwend, P. M., D. Backhus, J. K. MacFarlane, and A. L. Page. 1990. “Mobilization of
Colloids in Groundwater Due to Infiltration of Water at Coal Ash Disposal Site.” Journal of
Contaminant Hydrology, 6:307-320.
Gschwend, P. M., and M. C. Reynolds. 1987. “Monodispersed Ferrous Phosphate Colloids in an
Anoxic Groundwater Plume.” Journal of Contaminant Hydrology, 1:309-327.
Gschwend, P. M. and S. Wu. 1985. “On the Constancy of Sediment-Water Partition Coefficients
of Hydrophobic Organic Pollutants.” Environmental Science and Technology, 19:90-96.
Gu, B., and R. K. Schulz. 1991. Anion Retention in Soil: Possible Application to Reduce
Migration of Buried Technetium and Iodine. NUREG/CR-5464, U.S. Nuclear Regulatory
Commission, Washington, D.C.
Haggerty, R., and S. M. Gorelick. 1995. “Multiple-Rate Mass Transfer for Modeling Diffusion
and Surface Reactions in Media with Pore-scale Heterogeneity.” Water Resources Research,
31:2383-2400.
Hem, J. D. 1985. Study and Interpretation of the Chemical Characteristics of Natural Water.
Water Supply Paper 2254, U. S. Geological Survey, Alexandria, Virginia.
Higgins, G. H. 1959. “Evaluation of the Groundwater Contamination Hazard from Underground
Nuclear Explosives.” Journal of Geophysical Research, 64:1509-1519.
Hillel, D. 1998. Environmental Soil Physics. Academic Press, San Diego, California.
6.10
Ho, C. H., and N. H. Miller. 1986. “Formation of Uranium Oxide Sols in Bicarbonate
Solutions.” Journal of Colloid and Interface Science, 113:232-240.
Hollander, M., and D.A. Wolfe. 1973. Nonparametric Statistical Methods. Wiley, New York,
New York..
Honeyman, B. D., and P. H. Santschi. 1988. “Metals in Aquatic Systems.” Environmental
Science and Technology, 22:862-871.
Hopmans, J. W., H. Schukking, and P. J. J. F. Torfs. 1988. “Two-Dimensional Steady State
Unsaturated Water Flow in Heterogeneous Soils with Autocorrelated Soil Hydraulic
Properties.” Water Resources Research, 24:2005-2017.
INTERA (INTERA Environmental Consultants, Inc.). 1983. Geochemical Models Suitable for
Performance Assessment of Nuclear Waste Storage: Comparison of PHREEQE and
EQ3/EQ6. ONWI-473, INTERA Environmental Consultants, Inc., Houston, Texas.
Ivanovich, M., A. G. Latham, G. Longworth, and M. Gascoyne. 1992. “Applications to
Radioactive Waste Disposal Studies.” In Uranium-Series Disequilibrium. Applications to
Earth, Marine, and Environmental Systems, M. Ivanovich and R. S. Harmon (eds.),
pp. 583-630, Oxford University Press, Oxford, England.
Jackson, K. J., and W. L. Bourcier (technical organizers). 1986. Proceedings of the Workshop
on Geochemical Modeling. CONF-8609134, National Technical Information Service,
Springfield, Virginia.
Jackson, R. E., and K. J. Inch. 1989. “The In-situ Absorption of Sr-90 in a Sand Aquifer at the
Chalk River Nuclear Laboratories.” Journal of Contaminant Hydrology, 4: 27-50.
Jacobs, G. K., and S. K. Whatley (eds.). 1985. Proceedings of the Conference on the
Application of Geochemical Models to High-level Nuclear Waste Repository Assessment.
NUREG/CR-0062 (ORNL/TM-9585), Oak Ridge National Laboratory, Oak Ridge,
Tennessee.
James, R. V., and J. Rubin. 1986. “Transport of Chloride Ion in a Water-Unsaturated Soil
Exhibiting Anion Exclusion.” Soil Science Society of America Journal, 50:1142-1149.
Jardine, P. M., G. K. Jacobs, and G. V. Wilson. 1993. “Unsaturated Transport Processes in
Undisturbed Heterogeneous Porous Media: I. Inorganic Contaminants.” Soil Science
Society of America Journal, 57:945-953.
6.11
Jenne, E. A. 1977. “Trace Element Sorption by Sediments and Soils - Sites and Processes.” In
Symposium on Molybdenum in the Environment, W. Chappel and K. Petersen (eds.),
pp. 425-553, M. Dekker, Inc., New York, New York.
Jenne, E. A. (ed.). 1979. Chemical Modeling in Aqueous Systems. Speciation, Sorption,
Solubility, and Kinetics. Symposium Series 93, American Chemical Society,
Washington, D.C.
Jenne, E. A. 1981. Geochemical Modeling: A Review. PNL-3574, Pacific Northwest
Laboratory, Richland, Washington.
Jenne, E. A.. 1998a. “Adsorption of Metals by Geomedia: Data Analysis, Modeling, Controlling
Factors, and Related Issues.” In Adsorption of Metals by Geomedia. Variables,
Mechanisms, and Model Applications, E. A. Jenne (ed.), p. 2-73, Academic Press, San
Diego, California.
Jenne, E. A. 1998b. “Priorities for Future Metal Adsorption Research.” In Adsorption of Metals
by Geomedia. Variables, Mechanisms, and Model Applications, E. A. Jenne (ed.),
p. 549-560, Academic Press, San Diego, California.
Johnson, W. H., S. M. Serkiz, L. M. Johnson, and S. B. Clark. 1995. “Uranium Partitioning
Under Acidic Conditions in a Sandy Soil Aquifer.” Paper presented at the Waste
Management ‘95 Symposium, Tucson, Arizona, February 26 - March 2, 1995.
Jury, W. A., H. Elabd, and M. Resketo. 1986. “Field Study of Napropamide Movement Through
Unsaturated Soil.” Water Resources Research, 22:749-755.
Jury, W. A., W. R. Gardner, and W. H. Gardner. 1991. Soil Physics. John Wiley and Sons, Inc.
New York, New York.
Kaplan, D. I., P. M. Bertsch, and D. C. Adriano. 1995a. “Enhanced Transport of Contaminant
Metals Through an Acidic Aquifer.” Ground Water, 33:708-717.
Kaplan, D. I., P. M. Bertsch, D. C. Adriano, and W. P. Miller. 1993. “Soil-Borne Mobile
Colloids as Influenced by Water Flow and Organic Carbon.” Environmental Science and
Technology, 27:1193-1200.
6.12
Kaplan, D. I., P. M. Bertsch, D. C. Adriano, and K. A. Orlandini. 1994a. “Actinide Association
with Groundwater Colloids in a Coastal Plain Aquifer.” Radiochimica Acta, 66/67:181-187.
Kaplan, D. I., D. B. Hunter, P. M. Bertsch, S. Bajt, and D. C. Adriano. 1994b. “Application of
Synchrotron X-Ray Fluorescence Spectroscopy and Energy Dispersive X-Ray Analysis to
Identify Contaminant Metals on Groundwater Colloids.” Environmental Science &
Technology, 28:1186-1189.
Kaplan, D. I., T. L. Gervais, and K. M. Krupka. 1998. “Uranium(VI) Sorption to Sediments
Under High pH and Ionic Strength Conditions.” Radiochimica Acta, 80:201-211.
Kaplan, D. I., and R. J. Serne. 1995. Distribution Coefficient Values Describing Iodine,
Neptunium, Selenium, Technetium, and Uranium Sorption to Hanford Sediments.
PNL-10379, Supplement 1, Pacific Northwest Laboratory, Richland, Washington.
Kaplan, D. I., R. J. Serne, and M. G. Piepho. 1995b. Geochemical Factors Affecting
Radionuclide Transport Through Near and Far Fields at a Low-Level Waste Disposal Site.
PNL-10379, Pacific Northwest National Laboratory, Richland, Washington.
Kaplan, D. I., M. E. Sumner, P. M. Bertsch, and D. C. Adriano. 1996. “Chemical Conditions
Conducive to the Release of Mobile Colloids from Ultisol Profiles.” Soil Science Society of
America Journal, 60:269-274.
Karickhoff, S. W., D. S. Brown, and T. A. Scott. 1979. “Sorption of Hydrophobic Pollutants on
Natural Sediments.” Water Research, 13:231-248.
Kim, J. J. 1986. “Chemical Behavior of Transuranic Elements in Aquatic Systems.” In
Handbook on the Physics and Chemistry of the Actinides, A. J. Freeman and C. Keller (eds.),
pp. 413-455, Elsevier Science Publ., Amsterdam, Netherlands.
Kincaid, C. T., J. R. Morrey, and J. E. Rogers. 1984. Geohydrochemical Models for Solute
Migration. Volume 1: Process Description and Computer Code Selection. EPRI EA-3417,
Volume 1, Battelle, Pacific Northwest Laboratories, Richland, Washington.
Knoll, K. C. 1960. Adsorption of Strontium by Soils Under Saturated and Unsaturated Flow
Conditions. HW-67830, General Electric Company, Hanford Atomic Products Operation,
Richland, Washington.
Koltermann, C. E., and S. M. Gorelick. 1996. “Heterogeneity in Sedimentary Deposits:
A Review of Structure-Imitating, Process-Imitating, and Descriptive Approaches.” Water
Resources Research, 32:2617-2658.
6.13
Krupka, K. M., E. A. Jenne, and W. J. Deutsch. 1983. Validation of the WATEQ4
Geochemical Model for Uranium. PNL-4333, Pacific Northwest Laboratory, Richland,
Washington.
Krupka, K. M. and J. R. Morrey. 1985. MINTEQ Geochemical Reaction Code: Status and
Applications. In Proceedings of the Conference on the Application of Geochemical Models
to High-level Nuclear Waste Repository Assessment, G. K. Jacobs and S. K. Whatley (eds.),
pp. 46-53, NUREG/CR-0062 (ORNL/TM-9585), Oak Ridge National Laboratory, Oak
Ridge, Tennessee.
Krupka, K. M. and R. J. Serne. 1998. Effects on Radionuclide Concentrations by
Cement/Ground-Water Interactions in Support of Performance Assessment of Low-Level
Radioactive Waste Disposal Facilities. NUREG/CR-6377 (PNNL-11408), Pacific Northwest
National Laboratory, Richland, Washington.
LaGrega, M.D. 1994. Hazardous Waste Management. McGraw-Hill, Inc., New York,
New York.
Landström, O., C. E. Klockars, O. Persson, K. Andersson, B. Torstenfelt, B. Allard,
S. Å. Larsson, and E. L. Tullborg. 1982. “A Comparison of In-situ Radionuclide Migration
Studies in the Studsvik Area and Laboratory Measurements.” In Scientific Basis for Nuclear
Waste Management V, W. Lutze (ed.), pp. 697-706, Materials Research Society Symposia
Proceedings, Volume 11, North Holland, New York, New York.
Langmuir, D. 1978. “Uranium Solution-Mineral Equilibria at Low Temperatures with
Applications to Sedimentary Ore Deposits.” Geochimica et Cosmochimica Acta, 42:547-569.
Langmuir, D. 1997. Aqueous Environmental Geochemistry. Prentice Hall, Upper Saddle River,
New Jersey.
Langmuir, D., and J. S. Herman. 1980. “The Mobility of Thorium in Natural Waters at Low
Temperatures.” Geochimica Cosmochimica Acta 44:1753-1766.
Langmuir, I. 1918. “The Adsorption of Gases on Plane Surfaces of Glass, Mica, and Platinum.”
Journal of the American Chemical Society, 40:1361-1403.
Lehninger, A. L. 1970. Biochemistry. Worth Publishers, Inc., New York, New York.
Lemire, R. J., and P. R. Tremaine. 1980. “Uranium and Plutonium Equilibria in Aqueous
Solutions to 200°C.” Journal of Chemical and Engineering Data, 25:361-370.
Lewis, G. N., and M. Randall. 1961. Thermodynamics. Revised by K. S. Pitzer and L. Brewer,
McGraw-Hill Book Company, New York, New York.
6.14
Lewis, R. E., T. T. Jarvis, M. R. Jarvis, and G. Whelan. 1994. Eielson Air Force Base Operable
Unit 2 Baseline Risk Assessment. PNL-8752, Pacific Northwest Laboratory, Richland,
Washington.
Liang, L., J. F. McCarthy, L W. Jolley, J. A. McNabb, and T. L. Mehlhorn. 1993. “Iron
Dynamics: Observations of Transformation During Injection of Natural Organic Matter in a
Sandy Aquifer.” Geochimica et Cosmochimica Acta, 57:1987-1999.
Lindenmeier, C. W., R. J. Serne, J. L. Conca, A. T. Owen, and M. I. Wood. 1995. Solid Waste
Leach Characteristics and Contaminant-Sediment Interactions Volume 2: Contaminant
Transport Under Unsaturated Moisture Contents. PNL-10722, Pacific Northwest
Laboratory, Richland, Washington.
Lindsay, W. L., 1979. Chemical Equilibria in Soils. John Wiley and Sons, New York,
New York.
Loeppert, R. H., A. P. Schwab, and S. Goldberg (eds.). 1995. Chemical Equilibrium and
Reaction Models. Special Publication Number 42, Soil Science Society of America, Inc.,
Madison, Wisconsin.
Loux, N. T., D. S. Brown, C. R. Chafin, J. D. Allison, and S. M. Hassan. 1989. “Chemical
Speciation and Competitive Cationic Partitioning on a Sandy Aquifer Material.” Journal of
Chemical Speciation and Bioavailability, 1:111-125.
Lyman, W. J., W. F. Reehl, and D. H. Rosenblatt. 1982. Handbook of Chemical Property
Estimation Methods -- Environmental Behavior of Organic Compounds. McGraw-Hill, New
York, New York.
Mackay, D. M., W. P. Ball, and M. G. Durant. 1986. “Variability of Aquifer Sorption Properties
in a Field Experiment on Groundwater Transport of Organic Solutes: Methods and
Preliminary Results.” Journal of Contaminant Hydrology, 1:119-132.
McCarthy, J. F., and D. Degueldre. 1993. “Sampling and Characterization of Colloids and
Particles in Groundwater for Studying their Role in Contaminant Transport.” In
Environmental Particles, J. Buffle and J. P. van Leeuwen (eds.), pp. 247-315. Lewis
Publishers, Boca Raton, Florida.
McDonald, M. G., and A. W. Harbaugh. 1988. A Modular Three-Dimensional Finite-
Difference Groundwater Flow Model. U.S. Geological Survey Techniques of Water
Resources Investigations, Book 6, Chapter A1, U.S. Geological Survey, Reston, Virginia.
6.15
McKinley, I. G., and W. R. Alexander. 1993. “Assessment of Radionuclide Retardation: Uses
and Abuses of Natural Analogue Studies.” Journal of Contaminant Hydrology, 13:249-259.
McKinley. J. P., and E. A. Jenne. 1991. <Experimental Investigation and Review of the “Solids
Concentration” Effect in Adsorption Studies.’ Environmental Science and Technology,
25:2082-2087.
McMahon, M. A., and J. W. Thomas. 1974. “Chloride and Tritiated Water Flow in Disturbed
and Undisturbed Soils Cores.” Proceedings Soil Science Society of America, 38:727-732.
Meier, H., E. Zimmerhacki, G. Zeitler, P. Menge, and W. Hecker. 1987. “Influence of
Liquid/Solid Ratios in Radionuclide Migration Studies.” Journal of Radioanalytical and
Nuclear Chemistry, 109:139-151.
Melchior, D. C., and R. L. Bassett (eds.). 1990. Chemical Modeling in Aqueous Systems II.
Symposium Series 416, American Chemical Society, Washington, D.C.
Mercer, J. W., C. R. Faust, W. J. Miller, and F. J. Pearson, Jr. 1981. Review of Simulation
Techniques for Aquifer Thermal Energy Storage (ATES). PNL-3769, Pacific Northwest
Laboratory, Richland, Washington.
Mills, W. B., S. Liu, and F. K. Fong. 1991. “Literature Review and Model (COMET) for
Colloid/Metals Transport in Porous Media.” Ground Water, 29:199-208.
Morel, F. M. M. 1983. Principles of Aquatic Chemistry. John Wiley and Sons, New York, New
York.
Morel, F. M. M., and J. G. Hering. 1993. Principles and Applications of Aquatic Chemistry.
John Wiley and Sons, Inc., New York, New York.
Morel, F. M. M., J. G. Yeasted, and J. C. Westall. 1981. “Adsorption Models:
A Mathematical Analysis in the Framework of General Equilibrium Calculations.” In
Adsorption of Inorganics at Solid-Liquid Interfaces, M. A. Anderson and A. J. Rubin (eds.),
pp. 263-294, Ann Arbor Science Publishers, Inc., Ann Arbor, Michigan.
Morrey, J. R., C. T. Kincaid, and C. J. Hostetler. 1986. Geohydrochemical Models for Solute
Migration. Volume 3: Evaluation of Selected Computer Codes. EPRI EA-3417, Volume 3,
Battelle, Pacific Northwest Laboratories, Richland, Washington.
Mucciardi, A. N., I. J. Booker, E. C. Orr, and D. Cleveland. 1979. “Statistical Investigation of
the Mechanics Controlling Radionuclide Sorption, Part II.” In Second Contractor
Information Meeting. Task 4. R. J. Serne (ed.), Vol. 2, pp 333-425, PNL-SA-7352, Pacific
Northwest Laboratory, Richland, Washington.
6.16
Mucciardi, A. N., T. C. Johnson, and J. Saunier. 1980. “Statistical Investigation of the
Mechanics Controlling Radionuclide Sorption, Part III.” In Third Contractor Information
Meeting. Task 4, J. F. Relyea (ed.), Vol. 1, pp. 1-75, PNL-SA-8571, Pacific Northwest
Laboratory, Richland, Washington.
Muller, A. B., D. Langmuir, and L. E. Duda. 1983. “The Formulation of an Integrated Physico-
chemical-Hydrologic Model for Predicting Waste Nuclide Retardation in Geologic Media.” In
Scientific Basis for Nuclear Waste Management VI, D. G. Brookins (ed.), pp. 547-564,
Materials Research Society Symposia Proceedings, Volume 15, North Holland, New York,
New York.
Nielsen, D. R., and J. W. Biggar. 1961. “Miscible Displacement in Soils: I. Experimental
Information.” Soil Science Society of America Journal, 25:1-5.
Nielsen, D. R., and J. W. Biggar. 1962. “Miscible Displacement in Soils: II. Behavior of
Tracers.” Proceedings Soil Science Society of America, 26:125-128.
Nielsen, D. R., J. W. Biggar, and K. T. Erh. 1973. “Spatial Variability of Field-Measured Soil-
Water Properties.” Hilgardia, 42:215-259.
Nielsen, D. R., M. T. van Genuchten, and J. W. Biggar. 1986. “Water Flow and Solute
Transport Processes in the Unsaturated Zone.” Water Resources Research, 22:895-1085.
Nordstrom, D. K., and J. W. Ball. 1984. “Chemical Models, Computer Programs and Metal
Complexation in Natural Waters.” In Complexation of Trace Metals in Natural Waters,
C. J. M. Kramer and J. C. Duinker (eds.), pp. 149-169, Martinus Nijhoff/Dr. J. W. Junk
Publishing Co., Netherlands.
Nordstrom, D. K., and J. L. Munoz. 1985. Geochemical Thermodynamics. The
Benjamin/Cummings Publishing Co., Inc., Menlo Park, California.
Nordstrom, D. K., L. N. Plummer, T. M. L. Wigley, T. J. Woley, J. W. Ball, E. A. Jenne, R. L.
Bassett, D. A. Crerar, T. M. Florence, B. Fritz, M. Hoffman, G. R. Holdren, Jr., G. M. Lafon,
S. V. Mattigod, R. E. McDuff, F. Morel, M. M. Reddy, G. Sposito, and J. Thrailkill. 1979.
“Comparison of Computerized Chemical Models for Equilibrium Calculations in Aqueous
Systems.” In Chemical Modeling in Aqueous Systems. Speciation, Sorption, Solubility, and
Kinetics, E. A. Jenne (ed.), pp. 857-892, American Chemical Society Symposium Series 93,
Washington, D.C.
6.17
O’Conner, D. J., and J. P. Connolly. 1980. “The Effect of Concentration of Adsorbing Solids on
the Partition Coefficient.” Water Research, 14:1517-1523.
Oscarson, D. W., and H. B. Hume. 1998. “Effect of Solid:Liquid Ratio on the Sorption of Sr
2+
and Cs
+
on Bentonite.” In Adsorption of Metals by Geomedia. Variables, Mechanisms, and
Model Applications, E. A. Jenne (ed.), p. 277-289, Academic Press, San Diego, California.
Papelis, C., K. F. Hayes, and J. O. Leckie. 1988. HYDRAQL: A Program for the Computation
of Chemical Equilibrium Composition of Aqueous Batch Systems Including Surface-
Complexation Modeling of Ion Adsorption at the Oxide/Solution Interface. Technical Report
No. 306, Department of Civil Engineering, Stanford University, Stanford, California.
Parsons, R. 1982. “Surface Properties of Oxides.” Journal of Electroanalytical Chemistry,
118:2-18.
Peck, A. J., R. J. Luxmoore, and J. L. Stolzy. 1977. “Effects of Spatial Variability of Soil
Hydraulic Properties in Water Budget Modeling.” Water Resources Research, 13:348-354.
Penrose, W. R., W. L. Plozer, W. H. Essington, D. M. Nelson, and K. A. Orlandini. 1990.
“Mobility of Pu and Am Through a Shallow Aquifer in a Semiarid Region.” Environmental
Science & Technology, 24:228-234.
Petersen, L. W., P. Moldrup, O. H. Jacobsen, and D. E. Rolston. 1996. “Relations Between
Specific Surface Area and Soil Physical and Chemical Properties.” Soil Science, 161:9-21.
Peterson, R. G., and L. D. Calvin. 1986. “Sampling.” In Methods of Soil Analysis, Part 1.
Physical and Mineralogical Methods, A. Klute (ed.), American Society of Agronomy, Inc.,
Madison, Wisconsin.
Peterson, S. R., C. J. Hostetler, W. J. Deutsch, and C. E. Cowan. 1987a. MINTEQ User's
Manual. NUREG/CR-4808 (PNL-6106), Pacific Northwest Laboratory, Richland,
Washington.
Peterson, S. R., W. J. Martin, and R. J. Serne. 1986. Predictive Geochemical Modeling of
Contaminant Concentrations in Laboratory Columns and in Plumes Migrating from
Uranium Mill Tailings Waste Impoundments. NUREG/CR-4520 (PNL-5788), prepared by
the Pacific Northwest Laboratory, Richland, Washington.
Peterson, S. R., B. E. Opitz, M. J. Graham, and L. E. Eary. 1987b. An Overview of the
Geochemical Code MINTEQ: Application to Performance Assessment for Low-Level
Wastes. PNL-6112, Pacific Northwest Laboratory, Richland, Washington.
6.18
Phillips, S. L., F. V. Hale, L. F. Silvester, and M. D. Siegel. 1988. Thermodynamic Tables for
Nuclear Waste Isolation. Aqueous Solutions Database. NUREG/CR-4864 (LBL-22860),
Volume 1, Lawrence Berkeley Laboratory, Lawrence, California.
Pickens, J. F., R. E. Jackson, K. J. Inch, and W. F. Merritt. 1981. “Measurement of Distribution
Coefficients Using a Radial Injection Dual-Tracer Test.” Water Resources Research, 17:529-
544.
Pignatello, J. J. 1989. “Sorption Dynamics of Organic Compounds in Soils and Sediments.” In
Reactions and Movement of Organic Chemicals in Soils, B. L. Sawhney and K. Brown
(eds.), pp. 45-80. SSSA Special Publication 22, Soil Science Society of America, Inc.,
Madison, Wisconsin..
Plackett, R. L., and J. P. Burman. 1946. “The Design of Optimum Multifactorial Experiments.”
Biometrika, 33:305-325.
Plummer, L. N., B. F. Jones, and A. H. Truesdell. 1976, WATEQF - A FORTRAN IV Version of
WATEQ, A Computer Program for Calculating Chemical Equilibrium of Natural Waters.
Water Resources Investigations WRI 76-13, U.S. Geological Survey, Reston, Virginia.
Potter, R. W., III. 1979. "Computer Modeling in Low Temperature Geochemistry." Reviews of
Geophysics and Space Physics, 17:850-860.
Powell, R. M., and R. W. Puls. 1993. “Passive Sampling of Groundwater Monitoring Wells
Without Purging: Multilevel Well Chemistry and Tracer Disappearance.” Journal of
Contaminant Hydrology, 12:51-77.
Puls, R. W. 1990. “Colloidal Consideration in Groundwater Sampling and Contaminant
Transport Predictions.” Nuclear Safety, 31:58-65.
Rai, D., A. R. Felmy, and J. L. Ryan. 1990. “Uranium(IV) Hydrolysis Constants and Solubility
Product of UO
2
·xH
2
O(am).” Inorganic Chemistry, 29:260-254.
Rai, D., and J. M. Zachara. 1984. Chemical Attenuation Rates, Coefficients, and Constants in
Leachate Migration. Volume 1: A Critical Review. EPRI EA-3356, Volume 1, Electric
Power Research Institute, Palo Alto, California.
Rancon, D. 1973. “The Behavior in Underground Environments of Uranium and Thorium
Discharged by the Nuclear Industry.” In Environmental Behavior of Radionuclides Released
in the Nuclear Industry. IAEA-SM-172/55, pp. 333-346, International Atomic Energy
Agency, Vienna, Austria.
6.19
Rard, J. A. 1983. Critical Review of the Chemistry and Thermodynamics of Technetium and
Some of its Inorganic Compounds and Aqueous Species. UCRL-53440, Lawrence Livermore
National Laboratory, Livermore, California.
Read, D, P. J. Hooker, M. Ivanovich, and A. E. Milodowski. 1991. “A Natural Analogue Study
of an Abandoned Uranium Mine in Cornwall, England.” Radiochimica Acta, 52/53:349-356.
Reichle, R., W. Kinzelbach, and H. Kinzelbach. 1998. “Effective Parameters in Heterogeneous
and Homogeneous Transport Models with Kinetic Sorption.” Water Resources Research,
34:583-594.
Relyea, J. F. 1982. “Theoretical and Experimental Considerations for the Use of the Column
Method for the Use of the Column Method for Determining Retardation Factors.”
Radioactive Waste Management and the Nuclear Fuel Cycle, 3(2):151-166.
Ritger, P. D., and N. J. Rose. 1968. Differential Equations with Applications. McGraw Hill,
New York, New York.
Robin, M. J. L., E. A. Sudicky, R. W. Gillham, and R. G. Kachanoski. 1991. “Spatial Variability
of Strontium Distribution Coefficients and Their Correlation with Hydraulic Conductivity in
the Canadian Forces Base Borden Aquifer.” Water Resources Research, 27:2619-2632.
Routson, R. C., and R. J. Serne. 1972. One-Dimensional Model of the Movement of Trace
Radioactive Solute Through Soil Columns - The PERCEL Model. BNWL-1718, Pacific
Northwest Laboratory, Richland, Washington.
Routson, R. C., G. S. Barney, R. H. Smith, C. H. Delegard, and L. Jensen. 1981. Fission
Product Sorption Parameters for Hanford 200-Area Sediment Types. RHO-ST-35, Rockwell
Hanford Operations, Richland, Washington.
Roy, W. R., I. G. Drapac, S. F. J. Chou, and R. A. Griffin. 1991. Batch-type Procedures for
Estimating Soil Adsorption of Chemicals. EPA/530-SW-87-006-F, Office of Solid Waste and
Emergency Response, U.S. Environmental Protection Agency, Washington, D.C.
RTI (Research Triangle Institute). 1994. Chemical Properties for Soil Screening Levels. Draft
Report. Prepared for the Environmental Protection Agency, Office of Emergency and
Remedial Response, Washington, D.C.
Russo, D. 1989a. “Field-Scale Transport of Interacting Solutes Through the Unsaturated Zone
2. Analysis of the Spatial Variability of the Field Response.” Water Resources Research,
25:2487-2495.
6.20
Russo, D. 1989b. “Field-Scale Transport of Interacting Solutes Through the Unsaturated Zone
1. Analysis of the Spatial Variability of the Transport Properties.” Water Resources
Research, 25:2475-2485.
Ryan, J. M., and P. M. Gschwend. 1990. “Colloid Mobilization in Two Atlantic Coastal Plain
Aquifers: Field Studies.” Water Resources Research, 26:307-322.
Salter, P. F., L. L. Ames, and J. E. McGarrah. 1981a. The Sorption Behavior of Selected
Radionuclides on Columbia River Basalts. RHO-BWI-LD-48, Rockwell Hanford
Operations, Richland, Washington.
Salter, P. F., L. L. Ames, and J. E. McGarrah. 1981b. Sorption of Selected Radionuclides on
Secondary Minerals Associated with the Columbia River Basalts. RHO-BWI-LD-43,
Rockwell Hanford Operations, Richland, Washington.
Schindler, P. W., and G. Sposito. 1991. “Surface Complexation at (Hydro)oxide Surfaces.” In
Interactions at the Soil Colloid-Soil Solution Interface, G. H. Bolt, M. F. DeBoodt, M. H. B.
Hayes, and M. B. Mc Bride (eds.), pp. 115-145, Kluwer Academic Press, New York,
New York.
Schwarzenbach, R. P., and J. Westall. 1981. “Transport of Nonpolar Organic Compounds from
Surface Water to Groundwater. Laboratory Sorption Studies.” Environmental Science and
Technology, 15:1360-1367.
Scott, H. D., R. E. Phillips, and R. F. Paetzold. 1974. “Diffusion of Herbicides in One Adsorbed
Phase.” Proceedings Soil Science Society of America, 38:558-562.
Seaman, J. C., P. M. Bertsch, and W. P. Miller. 1995. “Ionic Tracer Movement Through Highly
Weathered Sediments.” Journal of Contaminant Hydrology, 20:127-143.
Sehmel, G. A. 1989. Cyanide and Antimony Thermodynamic Database for the Aqueous Species
and Solids for the EPA-MINTEQ Geochemical Code. PNL-6835, Pacific Northwest
Laboratory, Richland, Washington.
Sergeyeva, E. I., A. A. Nikitin, I. L. Khodakovskiy, and G. B. Naumov. 1972. “Experimental
Investigation of Equilibria in the System UO
3
-CO
2
-H
2
O in 25-200
"
C Temperature Interval.”
Geochemistry International, 9:900-910.
Serkiz, S. M. And W. H. Johnson. 1994. Uranium Geochemistry in Soil and Groundwater at
the F and H Seepage Basins (U). EPD-SGS-94-307, Westinghouse Savannah River
Company, Savannah River Site, Aiken, South Carolina.
6.21
Serne, R. J., R. C. Arthur, and K. M. Krupka. 1990. Review of Geochemical Processes and
Codes for Assessment of Radionuclide Migration Potential at Commercial LLW Sites.
NUREG/CR-5548 (PNL-7285), Pacific Northwest Laboratory, Richland, Washington.
Serne, R. J., and A. B. Muller. 1987. “A Perspective on Adsorption of Radionuclides onto
Geologic Media.” In The Geological Disposal of High Level Radioactive Wastes,
D. G. Brookins (ed.), pp. 407-443, Theophrastus Publications, S.A., Athens, Greece.
Serne, R. J., and J. F. Relyea. 1981. “The Status of Radionuclide Sorption-Desorption Studies
Performed by the WRIT Program.” In The Technology of high-Level Nuclear Waste
Disposal, Volume 1, pp. 203-254, DOE/TIC-621, Technical Information Center,
U.S. Department of Energy, Oak, Ridge, Tennessee.
Serne, R. J., R. C. Routson, and D. A. Cochran. 1973. Experimental Methods for Obtaining
PERCOL Model Input Verification Data. BNWL-1721, Pacific Northwest Laboratory,
Richland, Washington.
Seth, Rajesh, Donald MacKay, and Jane Muncke. 1999. “Estimating the Organic Carbon Partition
Coefficient and its Variability for Hydrophobic Chemicals.” Environmental Science and
Technology, 33:2390-2394.
Sheppard, J. C., J. A. Kittrick, and T. L. Hart. 1976. Determination of Distribution Ratios and
Diffusion Coefficients of Neptunium, Americium, and Curium in Soil-Aquatic Environments.
RTO-221-T-12-2, Rockwell Hanford Operations, Richland, Washington.
Sheppard, S. C., M. I. Sheppard, and W. G. Evenden. 1990. “A Novel Method Used to Examine
Variation in Tc Sorption Among 34 Soils, Aerated and Anoxic.” Journal of Environmental
Radioactivity, 11:215.
Silva, R. J., G. Bidoglio, M. H. Rand, P. B. Robourch, H. Wanner, and I. Puigdomenech. 1995.
Chemical Thermodynamics Series, Volume 2: Chemical Thermodynamics of Americium.
Elsevier Science, New York, New York.
Smith, R. M., and A. E. Martell. 1976. Critical Stability Constants. Volume 4: Inorganic
Complexes. Plenum Press, New York, New York.
Smith, R. M., and A. E. Martell. 1995. “The Selection of Critical Stability Constants.” In
Chemical Equilibrium and Reaction Models, R. H. Loeppert, A. P. Schwab, and S. Goldberg
(eds.), Special Publication Number 42, Soil Science Society of America, Inc., Madison,
Wisconsin.
Smith, R. M., A. E. Martell, and R. J. Motekaitis. 1997. NIST Critically Selected Stability
Constants of Metal Complexes Database. Version 4. Users Guide. NIST Standard
6.22
Reference Database 46, National Institute of Standards and Technology, Gaithersburg,
Maryland.
Smith, R. W., and E. A. Jenne. 1991. “Recalculation, Evaluation, and Prediction of Surface
Complexation Constants for Metal Adsorption on Iron and Manganese Oxides.”
Environmental Science and Technology, 25:525-531.
Smith, R. W. and E. A. Jenne. 1992. “Response to ‘Comment on “Recalculation, Evaluation,
and Prediction of Surface Complexation Constants for Metal Adsorption on Iron and
Manganese Oxides.’” Environmental Science and Technology, 26:1253-1254.
Smith, R. W. and E. A. Jenne. 1991. “Recalculation, Evaluation, and Prediction of Surface
Complexation Constants for Metal Adsorption on Iron and Manganese Oxides.”
Environmental Science and Technology, 25:525-531.
Smith, R. W. and E. A. Jenne. 1988. Compilation, Evaluation, and Prediction of Trip-Layer
Model Constants for Ions on Fe(III) and Mn(IV) Hydrous Oxides. PNL-6754, Pacific
Northwest Laboratory, Richland, Washington.
Snedecor, G. W., and W. G. Cochran. 1967. Statistical Methods. Sixth Edition, Iowa State
University Press, Ames, Iowa.
Sposito, G. 1984. The Surface Chemistry of Soils. Oxford University Press, New York,
New York.
Sposito, G. 1989. The Chemistry of Soils. Oxford University Press, New York, New York.
Sposito, G. 1994. Chemical Equilibria and Kinetics in Soils. Oxford University Press, New
York, New York.
Sposito, G., and J. Coves. 1988. SOILCHEM: A Computer Program for the Calculation of
Chemical Speciation in Soils. Kearny Foundation for Soil Science, University of California,
Riverside, California.
SSSA (Soil Science Society of America). 1997. Glossary of Soil Science Terms. Soil Science
Society of America, Madison, Wisconsin.
Strenge, D. L., and S. R. Peterson. 1989. Chemical Data Bases for the Multimedia
Environmental Pollutant Assessment System (MEPAS): Version 1. PNL-7145, Pacific
Northwest Laboratory, Richland, Washington.
Stumm, W. (ed.). 1987. Aquatic Surface Chemistry. Chemical Processes at the Particle-Water
Interface. John Wiley and Sons, New York, New York.
6.23
Stumm, W., and J. J. Morgan. 1981. Aquatic Chemistry. An Introduction Emphasizing
Chemical Equilibria in Natural Waters. John Wiley and Sons, New York, New York.
Sudicky, E. A. 1986. “A Natural Gradient Experiment on Solute Transport in a Sand Aquifer:
Spatial Variability of Hydraulic Conductivity and its Role in the Dispersion Process.” Water
Resources Research, 22:2069-2082.
Sugita, F., R. W. Gillham, and C. Mase. 1995. “Pore Scale Variation in Retardation Factor as a
Cause of Nonideal Reactive Breakthrough Curves. 2. pore Network Analysis.” Water
Resources Research, 31:113-119.
Szecsody, J. E., A. Chilakapati, J. M. Zachara, and A. L. Garvin. 1998. “Influence of Iron Oxide
Inclusion Shape on Co II/III EDTA Reactive Transport Through Spatially Heterogeneous
Sediment.” Water Resources Research, 34:2501-2514.
Szecsody, J. E., J. M. Zachara, and P. L. Bruckhart. 1994. “Adsorption-dissolution Reactions
Affecting the Distribution and Stability of Co (II) EDTA in Iron Oxide-Coated Sand.”
Environmental Science and Technology, 28(9):1706-1716.
Szecsody, J. E., J. M. Zachara, A. Chilakapati, P. M. Jardine, and A. S. Ferrency. 1998.
“Importance of Flow and Particle-Scale Heterogeneity on Co II/III EDTA Reactive
Transport.” Journal of Hydrology, 209:112-136.
Tompson, A.F.B., A. L. Schafer, and R.W. Smith. 1996. “Impacts of Physical and Chemical
Heterogeneity on Contaminant Transport in a Sandy Porous Medium.” Water Resources
Research, 32:801-818.
Tompson, A. F. B. 1993. “Numerical Simulation of Chemical Migration in Physically and
Chemically Heterogeneous Porous Media.” Water Resources Research, 29:3709-3726.
Tompson, A. F. B., and L. W. Gelhar. 1990. “Numerical Simulation of Solute Transport in
Three-Dimensional Randomly Heterogeneous Porous Media.” Water Resources Research,
26:2541-2562.
Truesdell, A. H., and B. F. Jones. 1973. WATEQ, A Computer Program for Calculating
Chemical Equilibria of Natural Waters. USGS-WRD-73-007, U.S. Geological Survey,
Washington, D.C.
Truesdell, A. H., and B. F. Jones. 1974. “WATEQ, A Computer Program for Calculating
Chemical Equilibria of Natural Waters.” U.S. Geological Survey Journal of Research,
2:233-248.
6.24
Turner, D. R. 1991. Sorption Modeling for High-level Waste Performance Assessment:
A Literature Review. CNWRA 91-011, Center for Nuclear Waste Regulatory Analysis, San
Antonio, Texas.
Turner, D. R. 1993. Mechanistic Approaches to Radionuclide Sorption Modeling.
CNWRA 93-019, Center for Nuclear Waste Regulatory Analysis, San Antonio, Texas.
Turner, D. R. 1995. Uniform Approach to Surface Complexation Modeling of Radionuclide
Sorption. CNWRA 95-001, Center for Nuclear Waste Regulatory Analysis, San Antonio,
Texas.
Turner, D. R., T. Griffin, and T. B. Dietrich. 1993. “Radionuclide Sorption Modeling Using
the MINTEQA2 Speciation Code.” In Scientific Basis for Nuclear Waste Management XVI,
C. G. Interrante and R. T. Pabalan (eds.), pp. 783-789, Materials Research Society
Symposium Proceedings, Volume 294, Materials Research Society, Pittsburgh, Pennsylvania.
van Genuchten, M. Th., and W. J. Alves. 1982. Analytical Solutions of the One-Dimensional
Convective-Dispersive Solute Transport Equation. Technical Bulletin No. 1661, U.S.
Department of Agriculture, Washington, D.C.
van Genuchten, M. Th., and P. J. Wierenga. 1976. “Mass Transfer Studies in Sorbing Porous
Media. I. Analytical Solutions.” Soil Science Society of America Journal, 40:473-480.
van Genuchten, M. Th., and P. J. Wierenga. 1986. “Solute Dispersion Coefficients and
Retardation Factors.” In Methods of Soil Analysis -- Part 1: Physical and Mineralogical
Methods, A. Klute (ed.), Second Edition, Chapter 44, pp. 1025-1054, Soil Science Society of
America, Madison, Wisconsin,
Voice, T. C., C. P. Rice, and W. J. Weber. 1983. “Effect of Solids Concentration on the
Sorptive Partitioning of Hydrophobic Pollutants in Aquatic Systems.” Environmental Science
and Technology, 17:513-518.
Wagman, D. D., W. H. Evans, V. B. Parker, R. H. Shumm, I. Halow, S. M. Bailey, K. L.
Churney, and R. L. Nuttall. 1982. “The NBS Tables of Chemical Thermodynamic Properties.
Selected Values for Inorganic and C1 and C2 Organic Substances in SI Units.” Journal of
Physical and Chemical Reference Data 11(Supplement No. 2):1-392.
Waite, T. D., J. A. Davis, T. E. Payne, G. A. Waychunas, and N. Xu. 1994. “Uranium(VI)
Adsorption to Ferrihydrite: Application of a Surface Complexation Model.” Geochimica et
Cosmochimica Acta, 58(24):5465-5478.
Wanner, H, and I. Forest (eds.). 1992. Chemical Thermodynamics Series, Volume 1: Chemical
Thermodynamics of Uranium. (I. Grenthe, J. Fuger, R. J. M. Konings, R. J. Lemire, A. B.
6.25
Muller, C. Nguyen-Trung, and H. Wanner, review team), North-Holland, Elsevier Science
Publishing Company, Inc., New York, New York.
Warrick, A. W., and A. Amoozegar-Fard. 1979. “Infiltration and Drainage Calculation Using
Spatially Scaled Hydraulic Properties.” Water Resources Research, 15:1116-1120.
Warrick, A. W., and D. R. Nielsen. 1980. “Spatial Variability of Soil Physical Properties in the
Field.” In Applications of Soil Physics, D. Hillel (ed.), Chapter 13, pp. 319-345, Academic
Press, New York, New York.
Westall, J. C. 1986. “Reactions at the Oxide-Solution Interface: Chemical and Electrostatic
Models.” In Geochemical Processes at Mineral Surfaces, J. A. Davis and K. F. Hayes (eds.),
pp, 54-78, ACS Symposium Series 323, American Chemical Society, Washington, D.C.
Westall, J. C. 1994. “Modeling of the Association of Metal Ions With Heterogeneous
Environmental Sorbents.” In Scientific Basis for Nuclear Waste Management Vol. XVIII.
Part 2, T. Murakami and R. C. Ewing (eds.), pp. 937-950, Materials Research Society
Symposia Proceedings, Volume 353, Materials Research Society, Pittsburgh, Pennsylvania.
Westall, J. C., and H. Hohl. 1980. “A Comparison of Electrostatic Models for the
Oxide/Solution Interface.” Advances in Colloid and Interface Science, 12:265-294.
Westall, J. C., J. L. Zachary, and F. M. M. Morel. 1976. MINEQL, A Computer Program for the
Calculation of Chemical Equilibrium Composition of Aqueous Systems. Technical Note 18,
Department of Civil Engineering, Massachusetts Institute of Technology, Cambridge,
Massachusetts.
Whelan, G. 1996. Use of Distribution Coefficients in Transport Modeling and Risk
Assessments. Letter Report, Pacific Northwest National Laboratory, Richland, Washington.
Whelan, G., J. W. Buck, D. L. Strenge, J. G. Droppo, Jr., B. L. Hoopes, and R. J. Aiken. 1992.
“An Overview of the Multimedia Assessment Methodology MEPAS.” Hazardous Waste and
Hazardous Materials, 9(2):191-208.
Whelan, G., J. P. McDonald, and C. Sato. 1996. Multimedia Environmental Pollutant
Assessment System (MEPAS): Groundwater Pathway Formulations. PNNL-10907, Pacific
Northwest National Laboratory, Richland, Washington.
Whelan, G., D. L. Strenge, J. G. Droppo, Jr., B. L. Steelman, and J. W. Buck. 1987. The
Remedial Action Priority System (RAPS): Mathematical Formulations. PNL-6200, Pacific
Northwest Laboratory, Richland, Washington.
6.26
Wildung, R. E., T. R. Garland and D. A. Cataldo. 1977. “Accumulation of Technetium by
Plants.” Health Physics, 32:314-317.
Wood, W. W., and J. Petratis. 1984. “Origin and Distribution of Carbon Dioxide in the
Unsaturated Zone of the Southern High Plains of Texas.” Water Resources Research,
20:1193-1208.
Yariv, S., and H. Cross. 1979. Geochemistry of Colloid Systems. Springer-Verlag, New York,
New York.
Yeh, G. T., and Y. Tsai. 1976. “Analytical Three-Dimensional Transient Modeling of Effluent
Discharges.” Water Resources Research, 12:533-540.
Yeh, G. T. 1981. AT123D: Analytical Transient One, Two, and Three Dimensional Simulation
of Waste Transport in the Aquifer System. ORNL-5602, Oak Ridge National Laboratory,
Oak Ridge, Tennessee.
APPENDIX A
Acronyms, Abbreviations, Symbols, and Notation
A.2
Appendix A
Acronyms, Abbreviations, Symbols, and Notation
A.1.0 Acronyms And Abbreviations
AA Atomic absorption
ASCII American Standard Code for Information Interchange
ASTM American Society for Testing and Materials
CCM Constant capacitance (adsorption) model
CDTA Trans-1,2-diaminocyclohexane tetra-acetic acid
CEAM Center for Exposure Assessment Modeling at EPA’s Environmental Research
Laboratory in Athens, Georgia
CEC Cation exchange capacity
CERCLA Comprehensive Environmental Response, Compensation, and Liability Act
DLM Diffuse (double) layer (adsorption) model
DDLM Diffuse double layer (adsorption) model
DOE U.S. Department of Energy
DTPA Diethylenetriaminepentacetic acid
EDTA Ethylenediaminetriacetic acid
EDX Energy dispersive x-ray analysis
EPA U.S. Environmental Protection Agency
EPRI Electric Power Research Institute
HEDTA N-(2-hydroxyethyl) ethylenedinitrilotriacetic acid
HLW High level radioactive waste
IAEA International Atomic Energy Agency
ICP Inductively coupled plasma
ICP/MS Inductively coupled plasma/mass spectroscopy
IEP (or iep) Isoelectric point
LLNL Lawrence Livermore National Laboratory, U.S. DOE
LLW Low level radioactive waste
MCL Maximum Contaminant Level
MEPAS Multimedia Environmental Pollutant Assessment System
MS-DOS® Microsoft® disk operating system (Microsoft and MS-DOS are register
trademarks of Microsoft Corporation.)
NPL Superfund National Priorities List
NRC U.S. Nuclear Regulatory Commission
NWWA National Water Well Association
OERR Office of Remedial and Emergency Response, U.S. EPA
ORIA Office of Radiation and Indoor Air, U.S. EPA
OSWER Office of Solid Waste and Emergency Response, U.S. EPA
A.3
PC Personal computers operating under the MS-DOS® and Microsoft® Windows
operating systems (Microsoft® Windows is a trademark of Microsoft
Corporation.)
PNL Pacific Northwest Laboratory. In 1995, DOE formally changed the name of the
Pacific Northwest Laboratory to the Pacific Northwest National Laboratory.
PNNL Pacific Northwest National Laboratory, U.S. DOE
PZC Point of zero charge
RCRA Resource Conservation and Recovery Act
SCM Surface complexation model
SDMP NRC’s Site Decommissioning Management Plan
TDS Total dissolved solids
TLM Triple-layer adsorption model
UK United Kingdom (UK)
UK DoE United Kingdom Department of the Environment
UNSCEAR United Nations Scientific Committee on the Effects of Atomic Radiation

A.4
A.2.0 List of Symbols for the Elements and Corresponding Names
Symbol Element Symbol Element Symbol Element
Ac Actinium
Ag Silver
Al Aluminum
Am Americium
Ar Argon
As Arsenic
At Astatine
Au Gold
B Boron
Ba Barium
Be Beryllium
Bi Bismuth
Bk Berkelium
Br Bromine
C Carbon
Ca Calcium
Cb Columbium
Cd Cadmium
Ce Cerium
Cf Californium
Cl Chlorine
Cm Curium
Co Cobalt
Cr Chromium
Cs Cesium
Cu Copper
Dy Dysprosium
Er Erbium
Es Einsteinium
Eu Europium
F Fluorine
Fe Iron
Fm Fermium
Fr Francium
Ga Gallium
Gd Gadolinium
Ge Germanium
H Hydrogen
He Helium
Hf Hafnium
Hg Mercury
Ho Holmium
I Iodine
In Indium
Ir Iridium
K Potassium
Kr Krypton
La Lanthanum
Li Lithium
Lu Lutetium
Lw Lawrencium
Md Mendelevium
Mg Magnesium
Mn Manganese
Mo Molybdenum
N Nitrogen
Na Sodium
Nb Niobium
Nd Neodymium
Ne Neon
Ni Nickel
No Nobelium
Np Neptunium
O Oxygen
Os Osmium
P Phosphorus
Pa Protactinium
Pb Lead
Pd Palladium
Pm Promethium
Po Polonium
Pr Praseodymium
Pt Platinum
Pu Plutonium
Ra Radium
Rb Rubidium
Re Rhenium
Rh Rhodium
Rn Radon
Ru Ruthenium
S Sulfur
Sb Antimony
Sc Scandium
Se Selenium
Si Silicon
Sm Samarium
Sn Tin
Sr Strontium
Ta Tantalum
Tb Terbium
Tc Technetium
Te Tellurium
Th Thorium
Ti Titanium
Tl Thallium
Tm Thulium
U Uranium
V Vanadium
W Tungsten
W Wolfram
Xe Xenon
Y Yttrium
Yb Ytterbium
Zn Zinc
Zr Zirconium
A.5
A.3.0 List of Symbols and Notation
" Dispersivity in the x, y, or z direction
"N Capacity factor or ratio of the moles per unit volume of water-saturated solid,
C
s
, to the moles per unit volume of liquid, C
l
( Activity coefficient
* Constrictivity of the porous media
*' Mass-related constant
, Parameter in Dubinin-Radushkevich isotherm model equal to “RT 1n (1 + 1/C
i
)”
8 First-order degradation/decay coefficient
2 Volumetric water content
2
m
Volume fraction of water associated with the mobile domain
2
v
Total water content
2
vz
Moisture content in the vadose zone
µ Mobility
D
b
Bulk density
D
particle
Particle density
F Net charge associated with the surface of adsorbing mineral as conceptualized in
electrostatic adsorption models
F
d
Charge associated with the diffuse layer d of counterions as conceptualized in
electrostatic adsorption models
F
$
Charge associated with the $ layer as conceptualized in electrostatic adsorption
models
F
o
Charge associated with the o layer as conceptualized in electrostatic adsorption
models
F
s
Surface charge at the Stern layer
F
sd
Standard deviation associated with the Gaussian solution
J Tortuosity of the porous media
L
x
Pore velocity in direction x
N Porosity
N
,
Effective porosity
N
m
Mobile water fraction as defined by the ratio of the volume fraction of water
associated with the mobile domain, 2
m
, to the total water content, 2
v
R Electrical potential
R
d
Potential at the diffuse layer
R
o
Potential at the surface (plane o)
R
s
Potential at the Stern layer
A Concentration of free or unoccupied surface absorption site on a solid phase
ads Adsorption
A
i
Concentration of adsorbate (or species) I on the solid phase at equilibrium
A
m
Adsorption capacity of adsorbent per unit mass
am Amorphous
A.6
aq Aqueous
C Radioactivity of tracer on sediment
C Constant capacitance term
CEC Cation exchange capacity
C
i
Concentration of adsorbate (or species) I in solution at equilibrium
C
l
Moles per unit volume of liquid
C
om
Concentration of organic material
C
s
Moles per unit volume of water-saturated solid
C
T
Total mass at the site per total site volume
C
Tp
Total mass at the site per dry weight of soil
D Proportionality constant or diffusion coefficient
D* Dispersion coefficient in the x, y, and z directions adjusted for retardation with the
retardation factor
D
a
Apparent diffusion coefficient
D
e
Effective diffusion coefficient
D
i
Intrinsic diffusion coefficient
D
mech
Mechanical dispersion
D
mol
Molecular diffusion coefficient
D
p
Diffusion coefficient for a species within a porous media
D
x
Dispersion coefficient in direction x
e
-
Free electron
e
-RF/RT
Boltzmann factor
Eh Redox potential of an aqueous system relative to the standard hydrogen electrode
F Faraday constant, 23,060.9 cal/V·mol
f
oc
Fraction (w/w) of organic material in soil
ªG
f
°
,298
Gibbs free energy of formation at 298 K
ªG
f
°
,T
Gibbs free energy of formation at temperature T
ªG
r
°
,298
Gibbs free energy of reaction at 298 K
ªG
r
°
,T
Gibbs free energy of reaction at temperature T
3
H Tritium
H
1
Thickness of the vadose zone
h
m
Mixing-zone thickness
ªH
f
°
,298
Enthalpy (or heat) of formation at 298 K
ªH
f
°
,T
Enthalpy (or heat) of formation at temperature T
ªH
r
°
,298
Enthalpy (or heat) of reaction at 298 K
ªH
r
°
,T
Enthalpy (or heat) of reaction at temperature T
I Ionic strength
IAP Ion activity product
J
ix
Flux of species I in direction x
K A constant in the Langmuir, Freundlich and Dubinin-Radushkevich isotherm
models
K
DR
Concentration-based, conditional equilibrium constant calculated from Dubinin-
Radushkevich adsorption isotherm
A.7
K
d
Concentration-based partition (or distribution) coefficient
K
d
act
Activity-based partition coefficient
K
dis
Dissolution equilibrium constant
K
ex
Exchange reaction constant
K
F
Concentration-based, conditional equilibrium constant calculated from Freundlich
adsorption isotherm
K
F
act
Activity-based, conditional equilibrium constant calculated from Freundlich
adsorption isotherm
K
L
Concentration-based, conditional equilibrium constant calculated from Langmuir
adsorption isotherm
K
L
act
Activity-based, conditional equilibrium constant calculated from Langmuir
adsorption isotherm
K
oc
Organic-carbon partition coefficient
K
om
Organic-matter partition coefficient
K
r,298
Equilibrium constant at 298 K
K
r,T
Equilibrium constant at temperature T
K
sp,T
Solubility product
l Liter
M Generic term for metal or radionuclide constituent
m Meter
M
A
Instantaneous mass released per unit area
M
ads
Mass of constituent I associated with the adsorbed phase in the vadose zone
M
aq
Mass of constituent I associated with the aqueous phase in the vadose zone
M
rel
Released mass
M
saturated
Total mass of constituent I associated with the saturated zone
M
sed
Sediment mass
M
Total
Total combined mass of constituent I in the vadose and saturated zones
M
vadose
Total mass of constituent I associated with the vadose zone
ml Milliliter
mol Mole
mV Millivolt
N Constant in the Freundlich isotherm model
n Total porosity
n
e
Effective porosity
pE Negative common logarithm of the free-electron activity
pH Negative logarithm of the hydrogen ion activity
pH
zpc
pH for zero point of charge
R Ideal gas constant, 1.9872 cal/mol·K
R
f
Retardation factor
s Solid phase species
SI Saturation index, as defined by log (IAP/K
r,T
)
SOH Unreacted surface site occupied by a hydroxyl group
A.8
SOH·M Used in the non-electrostatic adsorption models for an adsorption site occupied by
component M or surface-bound metal
SO·M Used in the electrostatic adsorption models for an adsorption site occupied by
component M or surface-bound metal
T Absolute temperature, usually in Kelvin unless otherwise specified
T
F
Total surface charge for plane o
t Time
t
max
End of the break-through curve during a column experiment
t
min
Beginning of the break-through curve during a column experiment
t
pulse
Mean residence time of a solute during a column experiment for a pulse release
t
T
Total advective travel time of the contaminant
t
ss
Mean residence time of a solute during a column experiment for a steady-state
release
t
step
Mean residence time for a step input/release
TDS Total dissolved solids
V
source
Volume associated with the contaminated source
V
w
Volume of water (or adsorbate solution)
v* Contaminant velocity
v
c
Contaminant velocity
v
d
Darcy velocity
v
p
Pore-water velocity
X
Gf
, Y
Gf
, Z
Gf
Green's functions (which are orthogonal) in the x, y, and z directions, respectively
x Distance in the x direction
y Off-centerline distance
Z Valence state
z Charge of ion
{ } Activity
[ ] Concentration
APPENDIX B
Definitions
B.2
CaX(s) +
90
Sr
2%
(aq) =
90
SrX(s) + Ca
2%
(aq)
Appendix B
Definitions
Absorption - partitioning of a dissolved species into a solid phase.
Adsorption - partitioning of a dissolved species onto a solid surface.
Adsorption Edge - the pH range where solute adsorption sharply changes from ~10% to ~90%.
Actinon - name occasionally used, especially in older documents, to refer to
219
Rn which forms
from the decay of actinium.
Activity - the effective concentration on an ion that determines its behavior to other ions with
which it might react. An activity of ion is equal to its concentration only in infinitely dilute
solutions. The activity of an ion is related to its analytical concentration by an activity
coefficient, (.
Alkali Metals - elements in the 1A Group in the periodic chart. These elements include lithium,
sodium, potassium, rubidium, cesium, and francium.
Alpha Particle - particle emitted from nucleus of atom during one type of radioactive decay.
Particle is positively charged and has two protons and two neutrons. Particle is physically
identical to the nucleus of the
4
He atom (Bates and Jackson 1980).
Alpha Recoil - displacement of an atom from its structural position, as in a mineral, resulting
from radioactive decay of the release an alpha particle from its parent isotope (e.g., alpha
decay of
222
Rn from
226
Ra).
Amphoteric Behavior - the ability of the aqueous complex or solid material to have a negative,
neutral, or positive charge.
Basis Species - see component species.
Cation Exchange - reversible adsorption reaction in which an aqueous species exchanges with an
adsorbed species. Cation exchange reactions are approximately stoichiometric and can be
written, for example, as
where X designates an exchange surface site.
B.3
Cation Exchange Capacity (CEC) - the sum total of exchangeable cations that a sediment can
adsorb.
Code Verification - test of the accuracy with which the subroutines of the computer code
perform the numerical calculations.
Colloid - any fine-grained material, sometimes limited to the particle-size range of <0.00024 mm
(i.e., smaller than clay size), that can be easily suspended (Bates and Jackson 1979). In its
original sense, the definition of a colloid included any fine-grained material that does not occur
in crystalline form.
Complexation (Complex Formation) - any combination of dissolved cations with molecules or
anions containing free pairs of electrons.
Component Species - “basis entities or building blocks from which all species in the system can
be built” (Allison et al. 1991). They are a set of linearly independent aqueous species in terms
of which all aqueous speciation, redox, mineral, and gaseous solubility reactions in the
MINTEQA2 thermodynamic database are written.
Detrital Mineral - “any mineral grain resulting from mechanical disintegration of parent rock”
(Bates and Jackson 1979).
Deuterium (D) - stable isotopes
2
H of hydrogen.
Disproportionation - is a chemical reaction in which a single compound serves as both oxidizing
and reducing agent and is thereby converted into more oxidized and a more reduced
derivatives (Sax and Lewis 1987). For the reaction to occur, conditions in the system must be
temporarily changed to favor this reaction (specifically, the primary energy barrier to the
reaction must be lowered). This is accomplished by a number of ways, such as adding heat or
microbes, or by radiolysis occurring. Examples of plutonium disproportionation reactions are:
3Pu
4+
+ 2H
2
O = 2Pu
3+
+ PuO
2
2+
+4H
+
3PuO
2
+
+ 4H
+
= Pu
3+
+ 2PuO
2
2+
+2H
2
O.
Electron Activity - unity for the standard hydrogen electrode.
Far Field - the portion of a contaminant plume that is far from the point source and whose
chemical composition is not significantly different from that of the uncontaminated portion of
the aquifer.
Fulvic Acids - breakdown products of cellulose from vascular plants (also see humic acids).
Fulvic acids are the alkaline-soluble portion which remains in solution at low pH and is of
B.4
lower molecular weight (Gascoyne 1982).
Humic Acids - breakdown products of cellulose from vascular plants (also see fulvic acids).
Humic acids are defined as the alkaline-soluble portion of the organic material (humus) which
precipitates from solution at low pH and are generally of high molecular weight (Gascoyne
1982).
Hydrolysis - a chemical reaction in which water reacts with another substance to form two or
more new substances. For example, the first hydrolysis reaction of U
4+
can be written as
U
4+
+ H
2
O = UOH
3+
+ H
+
.
Hydrolytic Species - an aqueous species formed from a hydrolysis reaction.
Ionic Potential - ratio (z/r) of the formal charge (z) to the ionic radius (r) of an ion.
Isoelectric Point (iep) - pH at which a mineral’s surface has a net surface charge of zero. More
precisely, it is the pH at which the particle is electrokinetically uncharged.
Lignite - a coal that is intermediate in coalification between peat and subbituminous coal.
Marl - an earthy substance containing 35-65% clay and 65-35% carbonate formed under marine
or freshwater conditions
Mass Transfer - transfer of mass between two or more phases that includes an aqueous solution,
such as the mass change resulting from the precipitation of a mineral or adsorption of a metal
on a mineral surface.
Mass Transport - time-dependent movement of one or more solutes during fluid flow.
Mire - a small piece of marshy, swampy, or boggy ground.
Model Validation - integrated test of the accuracy with which a geochemical model and its
thermodynamic database simulate actual chemical processes.
Monomeric Species - an aqueous species containing only one center cation (as compared to a
polymeric species).
Near Field - the portion of a contaminant plume that is near the point source and whose chemical
composition is significantly different from that of the uncontaminated portion of the aquifer.
Peat - an unconsolidated deposit of semicarbonized plant remains in a water saturated
environment.
B.5
Polynuclear Species - an aqueous species containing more than one central cation moiety, e.g.,
(UO
2
)
2
CO
3
(OH)
3
-
and Pb
4
(OH)
4
4+
.
Protium (H) - stable isotope
1
H of hydrogen.
Retrograde Solubility - solubility that decreases with increasing temperature, such as those of
calcite (CaCO
3
) and radon. The solubility of most compounds (e.g., salt, NaCl) increases with
increasing temperature.
Species - actual form in which a dissolved molecule or ion is present in solution.
Specific Adsorption - surface complexation via a strong bond to a mineral surface. For example,
several transition metals and actinides are specifically adsorbed to aluminum- and iron-oxide
minerals.
Sol - a homogeneous suspension or dispersion of colloidal matter in a fluid.
Solid-Solution Phase - a solid material in which a minor element is substituted for a major
element in a mineral structure.
Thoron - name occasionally used, especially in older documents, to refer to
220
Rn which forms
from the decay of thorium.
Tritium (T) - radioactive isotope
3
H of hydrogen.
Tritium Units - units sometimes used to report tritium concentrations. A tritium unit (TU) is
equivalent to 1 atom of
3
H (tritium) per 10
18
atoms of
1
H (protium). In natural water that
produces 7.2 x 10
-3
disintegrations per minute per milliliter (dpm/ml) of tritium, 1 TU is
approximately equal to 3.2 picocuries/milliliter (pCi/ml).
APPENDIX C
Standard Method Used At
Pacific Northwest National Laboratory
For Measuring Laboratory Batch K
d
Values

1
Relyea, J. F., R. J. Serne, and D. Rai. 1980. Methods for Determining Radionuclide
Retardation Factors: Status Report. Pacific Northwest Laboratory, Richland, Washington
C.2
Appendix C
Standard Method Used
At Pacific Northwest National Laboratory
For Measuring Laboratory Batch K
d
Values
The standard method reproduced below is used by the authors of this report and
their coworkers at the Pacific Northwest National Laboratory in Richland,
Washington for the measurement of K
d
values. It is adapted from the procedure
described in Relyea et al. (1980).
1
1.0 Applicability
This procedure describes the method for measuring radionuclide distribution coefficients (K
d
’s) of
geologic material. This procedure includes descriptions for analyses of unconsolidated, loosely
consolidated, consolidated porous, and intact, impermeable geological materials.
2.0 Definitions
• Cold wash: Contact of solid sample with nonradioactive groundwater for purposes of
establishing chemical equilibrium with nontracer aqueous constituents.
• Tracer: Radioactive element added to groundwater solution to indicate migration and
retardation events.
• Spiked groundwater: Groundwater with tracer.
• Blank tube: Centrifuge tube containing spiked groundwater but no solids.
• Radiation Work Procedure (RWP): This is a set of instructions for safe handling of
radioactive material in the laboratory. The RWP covers a number of topics and shall be
read and understood before performing any work in the laboratory.
3.0 Responsible Staff
• Task leader
• Cognizant staff
C.3
4.0 Procedure
4.1 Materials
pH meter
pH combination electrode 0-14 pH
Magnetic stirrer
Stir bars
Scintillation vials
pH buffers
Groundwater
No. 18 stainless steel sieve (1 mm)
No. 50 sieve (0.3 mm)
Mortar and pestle
Analytical balance (accuracy within ± 0.01 g) - Refer to operation manual specific
to balance for use instructions.
50 ml polycarbonate centrifuge tubes with screw caps
Teflon tape
Groundwater
Orbit shaker
Centrifuge
Vacuum pipets
0.45-micrometer polycarbonate membrane filters
Radioactive tracer
Plastic bags
4.2 Safety Precautions
In using radioactive substances and/or solutions protective clothing should be used to reduce
the possibility of contamination. Each laboratory is supplied with a radiation work procedure
(RWP) which outlines the types and quantities of radionuclides permitted with instructions
for handling. Record the number of the RWP in the laboratory record book.
4.3 Sample Characterization
Before the K
d
study perform the following analyses to characterize solid and groundwater
samples (perform groundwater analysis within one month prior to study). Include the
following:
C.4
4.3.1 For groundwater
• pH
• bulk chemistry (Ca, Mg, Na, K, Cl, NO
3
, SO
4
, CO
3
, HCO
3
)
4.3.2 For solids
• Mineralogy
• Surface area
• Cation exchange capacity
• Moisture content
• Particle size analysis
Procedures which may be used to determine the above parameters are referenced at the
end of this document. Record all results.
4.4 Sample Preparation
4.4.1 Groundwater
4.4.1.1 Filter groundwater through a 0.45-µm polycarbonate membrane before it
is used in a batch K
d
measurement.
4.4.1.2 If retardation parameters (such as pH, ionic strength, and complexing
ligand concentration) are to be studied, chemically analyze the synthetic
or altered groundwater after preparation and filtration and record results.
4.4.2 Solid
4.4.2.1 Unconsolidated Material. To remove particles greater than one
millimeter (>1.0 mm), the sample shall be wet-sieved with groundwater
by passing the sample through a No. 18 stainless steel sieve. If tests
with the material are to be conducted in an inert atmosphere or in a
controlled atmosphere, rock samples are to be prepared under those
same atmospheric conditions. This requires minimum contact of the
rock with air from the time it is removed from the earth until the time the
experiment is concluded. This condition holds for Sections 4.5.2.2 and
4.5.2.3. The particle size shall be determined and reported with results
from Section 4.3.
4.4.2.2 Loosely Consolidated Material. The sample shall be disaggregated by an
ultrasonic method or by hand with a mortar and pestle. A portion of the
intact material shall be preserved for dynamic testing. Disaggregation
C.5
shall proceed no farther than that required to reduce the sample to its
natural grain size. Fresh surfaces will be exposed to weathering, but this
procedure should reduce fracturing of particles to a minimum. Remove
particles >1.0 mm as in Section 4.5.2.1. The particle size distribution
after disaggregation shall be reported with results in Section 4.3.
4.4.2.3 Consolidated Porous Material (and intact, impermeable rock). A portion
of the intact sample shall be preserved (and maintained under conditions
that simulate those in situ) for dynamic testing. The remaining sample is
to be crushed to pass through a No. 18 sieve (<1 mm). Crushing must
be accomplished by means that minimize the introduction of extraneous
material, such as metal filings, into the sample. The sample should then
be wet sieved through a No. 50 sieve (0.30 mm) to obtain particle sizes
between 0.30 mm and 1.00 mm.
4.4.2.4 After samples have been sized (Sections 4.5.2.1, 4.5.2.2 or 4.5.2.3), they
must be homogenized to insure that the same particle size distribution is
obtained for each subsample to be studied.
4.4.3 Equilibrium
4.4.3.1 Prepare 50 ml polycarbonate centrifuge tubes with screw caps by
obtaining and recording tare weights and assigning identifications which
are unique to each sample tube.
4.4.3.2 After homogenizing, 1-g (1.0 g ± 0.01 g) samples are to be weighed
(and weights recorded) into centrifuge tubes. Wrap centrifuge tube
threads with Teflon tape to prevent leaks.
4.4.3.3 Thirty-milliliters of filtered, nonspiked (no radioactive tracer)
groundwater is added to each tube, including blanks with no soil, for a
“cold” wash. The tube caps are to be replaced before the tubes are
placed on a shaker for a gentle overnight agitation (about one oscillation
per second).
4.4.3.4 Next centrifuge the tubes to separate solids and liquids. Removed the
solutions with a vacuum pipettes to prevent removal of the rock sample
(some liquid will remain in the tube).
4.4.3.5 Repeated the wash procedure twice more for a total of three cold
(nonradioactive) washes. Before the centrifuge step on the third wash,
measure and record the pH of the solid-solution. If the pH has changed
from its natural equilibrium value as measured in the field, the rock
C.6
sample and groundwater have not yet re-established equilibrium.
Continue to wash until the pH is stable. A change in pH is most likely to
occur with samples of crushed rock (Section 4.4.2.3) because fresh
surfaces (either rock or cementing agents) have been exposed.
4.4.4 After removal of the third wash solution, each tube must be reweighed and the
weight must be recorded to determine the volume of excess solution left in each
sample. Secure the cap of each tube to prevent evaporation, which would result
in an increased salt concentration in the remaining solution. The excess solution
volume is found by dividing the excess solution weight by the solution density.
4.5 Addition of Tracer
4.5.1 The adding of tracer to a solution represents a critical step in the execution of
radionuclide migration studies. Two items must be carefully considered: (1) the
total amount of tracer added must be soluble in the volume of solution used and
(2) the chemical composition of the groundwater or synthetic groundwater must
remain unchanged, except for the addition of the radionuclide(s) to be studied.
4.5.2 Dry the tracers so that excess acid or base in the stock solution is removed. Do
not dry volatile tracers in acid media or they will be lost. The chemical produced
by drying must be soluble in the solutions used in experimentation. (An incorrect
procedure would be to dry plutonium basic media that would produce an
insoluble PuO
2
or Pu[OH]
4
precipitate.)
4.5.3 Exception to the dry-addition rule must be made in some cases for radionuclides
that have multiple oxidation states. When drying might change the tracer stock
solution’s oxidation state--such as Pu(VI) to Pu(IV)--tracer should be added to
solution in as small a volume as possible with as little excess salt and acid or base
as possible. Otherwise, a dry, soluble, salt-free tracer shall be added to
groundwater.
4.5.4 Allow the tracer solution to sit for at least one week under conditions to be used
in the experiment (in equilibrium with air if the aquifer is in equilibrium with air,
or under controlled atmosphere conditions if the aquifer is not in equilibrium with
air). Make any necessary adjustments to pH during the equilibration time.
Solution is to be filtered (0.45 µm) after equilibration prior to contact with the
geologic material.
4.5.5 Calculate and record the amount of tracer (mol/l) present in the groundwater just
prior to contact with the geologic material. Additionally, report any carrier
isotope of the element added with the tracer and any natural occurrence of the
element in groundwater.
C.7
4.6 Rock and Groundwater Contact
4.6.1 Thirty milliliters (30 ml) of filtered groundwater containing the radioactive tracer
is added to each sample tube containing one gram (1 g) of solid. In addition, 30
ml of spiked groundwater is placed in each of three empty (blank) centrifuge
tubes (prewashed as in Section 4.4.3). The blank tubes are needed to detect
sorption of tracer by centrifuge tube walls.
4.6.2 After replacing the tube caps, the tubes are placed in plastic bags (5 to 20 tubes
per bag) to contain any contamination caused by leaky tubes. Next, the tubes are
placed on a shaker (for linear reciprocating shaker, place tubes horizontally) so
that the solid-solution mixture makes maximum contact. Set the shaking speed
to 0.8 to 1.2 oscillations per second to ensure mixing of solid and liquid but to
reduce grinding of particles.
4.6.3 If time is not a parameter being studied, then contact between solid and liquid is
to be seven days (7 days). Record the actual contact time allowed. The samples
are then removed from the shaker and the tubes are visually checked for leaks
(decontaminate if necessary and discard leaky tubes).
4.6.4 The blank and sample tubes are centrifuged for twenty minutes (20 min) at
10,000 g (g = 980 cm/sec
2
) or more, and fifteen milliliters (15 ml) of effluent is
filtered through a pre-washed 0.45 µm polycarbonate-membrane-type filter.
(Pre-wash with groundwater from Section 2.2 to remove foreign particles and
soluble impurities). Analyze filtered effluent samples for tracer activity. Next,
the effluent is decanted from the blanks into cleanly washed tubes and the empty
blank tubes are analyzed for tracer activity adsorbed on tube walls. If tracer
activity on blank tube walls is greater than 10 percent of the total blank activity
(determined in Section 4.7.3), do not use the blank influent activity for K
d
calcu-
lation. If the activity sorbed on blank walls is significantly greater than
10 percent (using a one-tailed “t” test and combined counting error and statistical
variation between blanks), directly count the activity of the sample. Methods for
both cases follow.
5.0 Batch K
d
Calculations
5.1 When tracer is not sorbed by blank tube wall
5.1.1 Data needed for K
d
calculation are: (1) excess solution volume, V
excess
, (ml) left
from the third cold wash (weight of excess solution divided by solution density);
(2) mass of solid aquifer material, M
sed
, (g); (3) volume of groundwater with
radioactive tracer added, V
spike
(ml); (4) activity or concentration of tracer in the

C.8
A
i
' q
i
'
(C
blank
x V
spike
) & C
effluent
(V
spike
% V
excess
)
M
sed
(1)
K
d
'
(C
blank
x V
spike
) & C
effluent
(V
spike
% V
excess
)
C
effluent
x M
sed
(2)
K
d
'
C& (C
effluent
x V
excess
)
C
effluent
x M
sed
(3)
effluent solution, C
effluent
(dpm/ml); and (5) the tracer activity or concentration in
the influent blank, C
blank
, (dpm/ml).
5.1.2 The tracer concentration on the solid phase, A
i
(or q
i
), is:
The K
d
is then given by:
5.2 When tracer is sorbed by blank tube wall (Gamma or X-ray Emitting Isotopes)
5.2.1 If the radioactive tracer is adsorbed on the walls of blank tubes, determine the
tracer adsorbed by the solid by direct measurement. Use a traceable standard
made with the same type of geologic material as used in the test.
5.2.2 For this procedure, after the sediment and traced groundwater have contacted for
at least 7 days, the samples are centrifuged to separate solids from liquids, then
the liquid effluent is decanted from the sample tube using a vacuum pipette, and
the sample is weighed to determine the excess effluent solution volume (V
excess
).
The solid sample is then dried (it should be “air dried” in the same manner as
when originally weighed, either in air or in a controlled atmosphere) and
transferred to a clean polycarbonate centrifuge tube. The weight of the dry
sample M
sed
(g) is then determined and radiocounting of the dry sample is
performed for tracer activity, C (dpm), using the same detector, sample position,
and radioanalytical techniques as used for the attenuation standard prepared in
Section 5.2.1.
5.2.3 Determine the effluent tracer activity, C
effluent
(dpm/ml), in geometries that are
traceable to a standard. The K
d
can then be calculated from:
C.9
5.3 When tracer is sorbed by blank tube wall ("- or $-Emitting Isotopes)
5.3.1 If the radioactive tracer is adsorbed by sample container walls, only the effluent
activity can be determined simply and directly. Two options are available for
determination of the activity adsorbed by the rock sample. One method is to
remove both the solid sample and effluent from the original container and to strip
the isotope from the container wall by some means. Mass balance will allow
calculation of the K
d
if one knows the amount of radionuclide in the effluent on
the tube wall and the total radionuclide initially added. A second method is to
chemically remove the radionuclide from the rock sample and count it.
Problems with the first method include the possibility that some of the solid may
adhere to the wall and raise the apparent activity of the nuclide adsorbed by the
container. Removal of the solid sample may also cause leaching of the container
wall and result in an apparent low activity for nuclides adsorbed by the container.
This can be minimized by using tubes made of material most appropriate to your
sample; consider Teflon, glass, or various plastics to minimize adherence.
The second method is subject to incomplete removal of the nuclide from the solid
or loss of material during any additional steps required for extraction, or both.
6.0 Reporting Results from Radionuclide Migration Experiments
6.1 The following generic K
d
coding form (Table 1) includes the information to be obtained.
(The different data categories and abbreviations are described in Section 6.2.)
6.2 Explanation of K
d
Coding Form
6.2.1 Category I. Reference
A. Name of the person who performed experiments
B. Date that the experiment was started.
C. Comments regarding deviations from procedure, anomalies that occurred
during the process, other pertinent information.
C.10
Table 1. Generic K
d
coding form.
Reference
Experimental
Details
Geologic
Media
Aqueous
Phase
Nuclide
Adsorption
Function
A. Name A. Method A. Name A. BEG A. ISO A. K
d
B. Date Started B. State B. Origin B. Macro B. CONC B. Units
C. Comments C. Ratio C. Total C. Trace C. SPE C. Direction
D. Time D. Mineral D. END D. ADD D. NUM
E. Temperature E. CO
3
E. Loading
F. ATM F. OX
G. SEP G. CEC
H. Analyze H. AEC
I. RAD I. SA
6.2.2 Category II. Experimental Details
A. Method refers to batch, axial filter, column, intact core, channel
chromatography, and so forth. For batch method, add more detail as to
whether cold washes and blank corrections were used. For example, use
mnemonics such as
“BATCH (3W, BC) = batch, three cold washes, with blank tube sorption
correction”
“BATCH (OW) = batch, zero cold washes and no correction.”
B. State of geologic media such as crushed 40 µm; intact core 2.5 cm dia x 5
cm; tablet 1 cm x 0.5 cm; crushed 30-80 µm, etc.
C. Ratio of solids to solution for batch K
d
; for columns include pore velocity or
column velocity (for example, 1 PV = 1 cm/hr, CV = 0.5 cm/hr) and porosity
and column bulk density; PR = porosity, BD = bulk density.
D. Time of contact such as shaking time for batch system or residence time in
flow through columns (h) = hours, (d) = days.
C.11
E. Temp is the temperature of the experiment in EC.
F. ATM is the equilibrating atmosphere air, N
2
, Ar, 10 percent CO
2
- 90 percent
Ar, and so forth.
G. SEP stands for separation technique; did you use filters (give median pore
size) or centrifugation (include approximate g’s)?
“FIL(.4) = filter 0.4 m”
“CEN(50) = centrifuged at 50 g’s where g = 980 cm/sec
2
units.”
H. Analyze states whether the K
d
is determined by analyzing (or counting)
liquids only or solid and liquid:
“L/L = liquids only”
“S/L = solid and liquid”
I. RAD is a list of all radioisotopes that were run simultaneously in the
experiment. Example: “Sr, Cs, Tc” means these isotopes were run together.
6.2.3 Category III. Geologic Media
A. Name. Use the generic name of the rock or mineral, e.g., basalt, granite,
montmorillonite.
B. Origin. Include a geographic description and some formation information,
e.g., Eleana shale, Sentinel Gap basalt, Argillaceous Shale Wards #404561.
C. Total. Identify the chemical composition as oxides (SiO
2
, Al
2
O
3
TiO
2
, FeO,
Fe
2
O
3
, MnO, CaO, MgO, K
2
O, Na
2
O, P
2
O
5
in percent.
D. Minerals. Identify the minerals present in the rock sample, listing the major
ones first, the minor ones last, in the order of the composition percentages in
which they appear (largest first). If there are quantitative estimates, add this
information as percent and tr = 5 percent.
E. CO
3
= carbonate content of rock.
F. OX = hydrous Fe, Mn, Al oxides content of rock.
C.12
G. CEC = cation exchange content of material; units = meg/100g. Specify pH
of system (typically pH = 7).
H. AEC = anion exchange content of material; units = meq/100g. Specify pH of
system.
I. SA = surface area; use “EG” for ethylene glycol, “BET” for gas adsorption,
use units m
2
/g, for example: EG(1.3).
6.2.4 Category IV. Aqueous Phase
A. BEG signifies measurements made prior to tracer adsorption.
B. Macro constituents include:
· pH
· Eh (units vs. S.H.E.)
· Na
+
· Ca
2+
· K
+
· Mg
2+
· Cl
-
· HCO
3
-
; CO
3
2-
· SO
4
2-
· SiO
4
C. Trace constituents include:
· NO
3
, ppm
· Organic carbon
· B
· Trace metals or anything else measured.
D. END signifies measurements (if performed) taken at the same time as K
d
determined.
6.2.5 Category V. Nuclide
A. ISO. Isotope used such as
237
Pu,
95m
Tc.
B. CONC. Concentration added to groundwater in M = molarity. Include any
carrier if present.
C.13
C. SPE. Species or valence state added, if known. Also state whether the
valence state distribution was determined after equilibration state, e.g.,
“Pu(VI) BEG; Pu(IV) 15 percent, Pu(V) 50 percent, Pu(VI) 10 percent
END” (which means that the original spike wads 100 percent Pu(VI), and
after shaking the final distribution was as shown).
D. ADD describes how the tracer was added to the groundwater; DRY means
evaporated to dryness and groundwater added; WET/PH/3DFO.4 means a
small aliquot of liquid tracer was added to the groundwater, the pH of the
system was re-adjusted to the appropriate value and shaken for 3 days to
filtration through 0.4 µm filters before usage.
“DRY/1DC50” means the dried spike was brought back into solution
equilibrated for one day, and centrifuged at 50 g’s before usage.
E. Loading describes (a) the percent of total exchange capacity of the
adsorbent filled with the nuclide of interest or (b) the mass of nuclide
adsorbed/mass of adsorbent at the condition when the K
d
measurement is
performed. This value can be calculated from knowledge of the cation or
anion exchange capacity in case (a) and from mass balance considerations.
One must know the original mass of the nuclide used in each experiment.
6.2.6 Category VI. Adsorption Function
A K
d
. Place the value for K
d
. If a retardation factor is determined in a flow-
through column as a function of water velocity, designate by the symbol RF.
Where several measurements were made, also give the standard deviation,
such as
“75 ± 12 = a K
d
”
“(RF) 60 ± 30 = retardation factor”
B. Units. ml/g or ml/m
2
.
C. Direction. ADS = adsorption direction
DES = desorption direction
ADS-DES = A spike addition to a column.
D. NUM = number of observations used to derive data point, for example:
3 = triplicate samples.
